Research is paramount to the mission of the National MPS Society. Focusing on areas of unmet need and developing treatment options for all diseases remains at the forefront.


The 2024 reseach program will open with Cycle II Letters of Intent (LOI) due July 15, 2024
Submission Portal will be live for Cycle II-2024 approximately the middle of June, 2024. The decision to forego an open call for proposals as part of Cycle I-2024 involved logistical considerations only. The Soceity remains committed to supporting research at the highest level and with a similar level committement as in years past.
The National MPS Society’s Innovative Research Program uses Proposal Central for grant submission, review, award, managment, and reporting. Go to the Proposal Central site and search for National MPS Soociety to start your application! Individual investigators and institutions should be registered on the Proposal Central website:
https://proposalcentral.com/
To give researchers and reviewers both the certainty of a set calendar of grant submission dates and to allow multiple submission cycles per year, the Society has three grant proposal submission cycles per year. As the individual cycle proceeds, the details of a subsequent individual call for may be altered, or a call may be cancelled, pending the results of the previous cycle. The dates and additional details are below.

Timeline for Grant Cycle submission dates etc. Submissions are due by 11:59:59 PM EST/EDT (USA) of the due date. If a due date falls on a USA federal holiday or a Saturday or Sunday, the due date will shift to the next business day.
The Society Research Portal should be open three weeks before a LOI due date.
Questions may be addressed to either grants@mpssociety.org or directly to the Society CSO, Matthew Ellinwood (matthew@mpssociety.org).

December 2017
Progress Report for study entitled “Evaluation of adeno-associated virus gene therapy in the feline model of mucolipidosis II”
Principle Investigators:
Allison M. Bradbury, PhD of University of Pennsylvania
Charles H. Vite, DVM, PhD of University of Pennsylvania
Steven J. Gray, PhD of University of North Carolina
The combination of efficacy and safety data on intravenous AAV9 gene therapy from animal models and the preliminary data from human trials are highly encouraging. We therefore believe that intravenous delivery of AAV9 holds the strongest potential for effectively treating ML II and should be evaluated in the feline model of ML II. Aim 1 of this grant proposal entailed generation and preliminary characterization of AAV vector encoding feline GNPTAB. During this reporting period we have created two AAV9 vector constructs, AAV9-Cbh-GNPTABopt-sPa and AAV9-Jet+I-GNPTABopt-SV40pA, the first driving a stronger level of expression of the GNPTAB protein and the second a weaker level of expression. Two cohorts of wild type mice have now be treated intravenously with 1 of the constructs, AAV9-Cbh-GNPTABopt-sPa or AAV9-Jet+I-GNPTABopt-SV40pA, and are currently being evaluated for safety and toxicity. Once safety and toxicity studies in mice are completed, large-scale vector manufacturing will begin to treat MLII cats in the next reporting period.
SA Pathology (WHC site)
Adelaide, Australia
“Can the cell cycle be reset to normal in the MPS growth plate chondrocytes?”
Telethon Institute of Genetics and Medicine (TIGEM)
Pozzuoli, Italy
“Disease mechanisms leading to dopaminergic dysfunction underlying behavioral symptoms in MPS IIIA”
Georgetown University
Washington, D.C.
“The Effects of Tyrosine Kinase Inhibition on MPS IIIA Mice”
University of Minnesota
Minneapolis, Minnesota
“Neurocognitive and neuroimaging of Morquio Syndrome – MPS IV”
Department of Pediatrics, University of Minnesota
Minneapolis, Minnesota
“Probing Oxidative Stress and Neuroinflammation as Potential Therapeutic Targets in MPS I”
Clinical Division of Endocrinology and Metabolism, Medical University of Vienna
Vienna, Austria
“Metabolic, microstructural and functional hallmarks of brain alteration in Mucopolysaccharidosis type II.”
The National MPS Society allocated $335,000 in grant funding for 2016, which includes the second year funding for grants awarded in 2015. We received many letters of intent from researchers around the world for research grants. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from researchers. Two new grants were chosen for MPS IVA and for MPS VII. Additionally, $100,000 of funds raised were through the Million Dollar Bike Ride effort, and these funds have been allocated through the University of Pennsylvania for MPS research grants.
The Board of Directors allocated $30,000 through the Fundraising Directive Program. The family who raised these funds requested work be continued with Dr. Haiyan Fu of the Research Institute at Nationwide Children’s Hospital for gene therapy approach for advanced MPS II via AAV9 vectors.
We also provided $25,000 to support the Lysosomal Disease Network’s NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. The MPS Society also provided $5,000 in partnership with the Ryan Foundation for an MPS I project.
In the fourth quarter of 2016, we raised an additional $150,000 in research funds to be awarded for two new projects in the first quarter of 2017. $135,000 will be awarded to Dr. Charbel Moussa of Georgetown University for an MPS IIIA drug repurposing project and $30,000 will be awarded in a partnership grant with the ISMRD (International Society for Mannosidosis and Related Diseases) for novel ML gene therapy research with Dr. Stephen Gray at UNC Hospital in partnership with Dr. Charles Vite at the University of Pennsylvania.
To evaluate AAV Gene Therapy in the feline model of ML II
Dr. Allison Bradbury, University of Pennsylvania
Dr. Charles Vite, University of Pennsylvania
Dr. Steven Gray, University of North Carolina at Chapel Hill
Dr. Kim Hemsley
South Australia Health and Medical Research Institute
Adelaide, South Australia
Dr. Kazuki Sawamoto, Dr. Shunji Tomatsu
Nemours/Alfred I. duPont Hospital for Children
Wilmington, DE
Therapeutic Targeting of Wnt/β-Catenin Signaling to Improve Bone Formation in MPS VII
Dr. Lachlan J. Smith
University of Pennsylvania
Philadelphia, PA
Targeting mTORCI and autophagy pathways to rescue the skeletal phenotype in MPS mouse models
Dr. Carmine Settembre
Telethon Institute of Genetics and Medicine of Fondazione Telethon
Pozzuoli, Italy
Validation of small molecule therapeutic leads for treatment of MPS I disease
Dr. Allison R. Kermode, Professor
Simon Fraser University
Burnaby, BC Canada
Dr. Kim Hemsley, Dr. Alessandro Fraldi, and Professor Robert D. Jolly
Lysosomal Diseases Research Unit
Adelaide, SA, Australia
Creating new tools for understanding skeletal disease in MPS IVA
Dr. Ainslie Derrick-Roberts
Genetics and Molecular Pathology
North Adelaide, SA, Australia
Igor Nestrasil, MD
University of Minnesota
Mucopolysaccharidoses pilot for $50,500
Microstructural and functional MRI signatures in patients with MPS I
The National MPS Society allocated $509,000 in grant funding for 2014 which includes the second year funding for grants awarded in 2013, the unspent MPS III funds from 2012 and the 2014 grants. The funding we provide is critical as we move forward with our mission to find cures for MPS and ML. We received 20 letters of intent from researchers around the world for the General, MPS II, MPS IVA and MPS VI grants. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from eight researchers.
The board of directors allocated $100,000 to Abeona Therapeutics which has licensed the MPS IIIA and MPS IIIB gene therapy technology from Nationwide Children’s Hospital. Funds already raised by Abeona have been funneled to Nationwide for MPS III drug manufacturing and preclinical research plus two INDs (investigational new drug applications). The financial distribution from the Society will help move the clinical trial forward.
We also provided $25,000 to support the Lysosomal Disease Network’s NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. An additional $8,000 was offered for an ML grant in partnership with ISMRD (International Society for Mannosidosis and Related Diseases). A $10,000 partnership grant with the Ryan Foundation funds the University of MN project “Longitudinal Studies of Brain Structure and Function in MPS Disorders.” We also provide funding for post-doctoral fellows to attend the American Society and Gene and Cell Therapy conference.
Moin Vera, MD, PhD
Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center
Torrance, CA
AAV mediated gene transfer to the CNS for MPS II
Scott McIvor, PhD
University of Minnesota
Minneapolis, MN
Overcoming limitations inherent in sulfamidase to improve MPS IIIA gene therapy
Beverly Davidson, PhD
The Children’s Hospital of Philadelphia
Pentosan Polysulfate and GAGs in MPS
Icahn School of Medicine at Mount Sinai
New York, NY
The National MPS Society allocated $530,000 in grant funding for 2013, which includes the second year funding for grants awarded in 2012, plus the 2013 grants. The funding we provide is critical as we move forward with our mission to find cures for MPS and ML. We received 48 letters of intent from researchers around the world for the six grants offered in 2013. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from 13 researchers.
The MPS Society will also fund $25,000 to support the Lysosomal Disease Network’s NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. An additional $20,000 will be offered for an ML grant in partnership with ISMRD (International Society for Mannosidosis and Related Diseases). A $10,000 partnership grant with the Ryan Foundation funded the University of MN project “Longitudinal Studies of Brain Structure and Function in MPS Disorders.” The National MPS Society also provides funding for post-doctoral fellows to attend the Gordon Conference on lysosomal diseases.
Pathogenesis of Bone Disease in Mucopolysaccharidosis Disorders
two years @ $30,000 each year
Lachlan Smith, PhD
University of Pennsylvania
Philadelphia, PA
Adjunctive therapy for Hurler syndrome.
Richard Steet, PhD
University of Georgia
Athens, GA
AND
Dr. Dwight Koeberl
Duke University
Durham, NC
Development of pharmacological chaperone therapy for MPS II.
Vito Ferro, PhD
University of Queensland
Brisbane, Queensland
Australia
Delivery of sulfamidase to the brain.
Jeffrey Esko, PhD
University of California, San Diego
La Jolla, CA
Manifestations of Cardiovascular Disease in Morquio A: Evaluation, Assessment, and Therapy
Adriana Montano, PhD
St. Louis University
St. Louis, MO
AND
Raymond Wang, M.D.
CHOC Children’s Hospital
Orange, CA
The National MPS Society awarded $547,000 in grant funding for 2012 which includes the second year funding for grants awarded in 2011 plus the 2012 grants. The funding we provide is critical as we move forward with our mission to find cures for MPS and ML. We received 16 letters of intent from researchers around the world for the three grants offered in 2012. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from seven researchers.
We also provided $25,000 to support the Lysosomal Disease Network’s NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. An additional $15,000 has been allocated for a mucolipidosis partnership grant with the Gandhi Foundation to Dr. Sara Cathey at Greenwood Genetics Center, “PTC 124 for nonsense mutation suppression in ML II and III cultured fibroblasts.” A $10,000 partnership grant with the Ryan Foundation funded the University of MN project “Brain Structure and Function in Developmentally Normal Children Ages 4-7.” The National MPS Society also provides funding for post-doctoral fellows to attend scientific meetings, such as the American Society of Gene and Cell Therapy.
Gustavo H.B. Maegawa, MD, PhD
Johns Hopkins School of Medicine, Department of Pediatrics
Baltimore, MD
Development of Long Circulating Enzyme Replacement Therapy for MPS IVA.
Shunji Tomatsu, MD, PhD
Nemours Children’s Clinic – Delaware Valley of the Nemours Foundation
Wilmington, DE
Dr. Brian Bigger
Stem Cell & Neurotherapies Group
Manchester, UK
“Evaluation of high dose genistein aglycone in the treatment of mucopolysaccharide disease types IIIA, B and C.”
2011 Research Grant PDF Download
The National MPS Society awarded $421,500 in grant funding for 2011. The funding the Society provides has been and continues to be crucial as we move forward with our mission to find the cures.
We received 33 letters of intent from researchers around the world for the five grants offered in 2011. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from 13 researchers.
All grant recipients were awarded $70,000 for the two year grant, with half of the total provided each year. The Society will fund $25,000 to support the Lysosomal Disease Networks NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. The Society also provides funding for post-doctoral fellows to attend scientific meetings, such as the American Society of Gene and Cell Therapy and the Gordon Conference on Lysosomal Diseases.
MPS II is caused by defects in an enzyme called iduronate-2-sulfatase. L Many of these defects result in degradation of the enzyme in cells before it has had a chance to carry out its normal function, thus producing clinical symptoms. MPS II patients may be treated with enzyme replacement therapy in which a synthetic, fully functional enzyme is administered by injection. Unfortunately, the replacement enzyme cannot cross the blood-brain barrier and thus cannot relieve the neurological symptoms associated with severe cases of MPS II. The aims of this project are to develop small molecules for the treatment of MPS II, which unlike enzymes, are capable of crossing the blood-brain barrier and thus may offer relief of neurological symptoms. The small molecules are designed to act as chaperones to protect the defective enzyme from degradation and restore enzyme activity to sufficient levels to relieve symptoms. This approach has shown great promise in other lysosomal storage diseases but has yet to be extended to MPS II. This project will address that situation.
Animal models of MPS types I, II and IIIA can be treated by providing recombinant enzyme into the fluid surrounding the brain (cerebrospinal fluid). The application of this treatment of IIIB has been hampered by the inability of the missing enzyme, alpha-?-acetylglucosaminidase (NAGLU), to enter cells efficiently. We have created NAGLU tagged with insulin-like growth factor 2 (IGF2) which enters cells effectively using the mannose 6-phosphate receptor. Here, we will deliver NAGLU-IGF2 to the cerebrospinal fluid by using gene therapy in animal models. We will target the part of the brain that makes cerebrospinal fluid (the choroid plexus), to determine whether this will provide a source of NAGLU-IGF2 for the brain. This study will provide proof-of-principle for choroid-plexus directed gene therapy with NAGLU-IGF2 as a potential therapy for MPS III IIIB, to determine whether this approach can deliver enzyme using cerebrospinal fluid without the need for repeated injections.
Enzyme replacement therapy for MPS VI requires weekly administrations of costly enzyme and has a poor outcome on some of the disease characteristics including bone and cartilage abnormalities. We have recently shown that a single systemic delivery of an adeno-associated viral vector (AAV) encoding the correct copy of the enzyme missing in MPSVI results in: i.sustained expression of the enzyme from liver of MPS VI cats transduced by AAV; ii.significant amelioration of the disease phenotype (including bone and cartilage) in this large model of the disease.
Based on these promising results we are planning a clinical trial to test the safety and efficacy of our approach in MPS VI patients. Towards this goal we propose to:
We believe these data will be instrumental to rapidly move gene therapy for MPS VI from bench to bedside thus overcoming some of the limitations of current therapies. In addition, the results from these studies may improve the cures for other MPS.
Morquio A disease is characterized with the build-up of two specific sugars (chondroitin-6- sulfate and keratan sulfate) in all the body cells, particularly in skeletal tissue. Effects of this build-up on the immune system and the consequences on the cartilage destruction and alterations of bone metabolism in Morquio A disease have not been investigated yet. We will clarify the role of immune system in the pathogenesis of Morquio A disease. We will characterize the immune profile of Morquio A mouse bone and cartilage cells and tissues, as well as Morquio A human cartilage cells. The outcome of this research will enable us to develop better approaches for treatment strategies to stop cartilage degeneration not only in Morquio A but also in other MPS.
Understanding the molecular events that cause disease symptoms is an important step in the development of new therapies for many inherited disorders, especially in cases where replacement of the defective gene or enzyme is difficult. In earlier studies, we showed that the cartilage defects in a zebrafish model for ML-II are associated with increased activity of protein-degrading enzymes called cathepsins and excessive TGF-beta signaling. Our most recent work now demonstrates that reducing the activity of one of these enzymes, cathepsin K, results in correction of the cartilage defects in ML-II zebrafish embryos. Using known drugs, we now propose to block the activity of two other cathepsin proteases and to reduce excessive TGF-beta signaling to determine how these molecules impact the onset and progression of disease phenotypes such as impaired development of cartilage and heart valves. Since elevated cathepsin activity is a common feature of many MPS disorders, we believe our results on ML-II will increase our understanding of the disease mechanisms associated with other lysosomal diseases.
The aim of this project is the development of small molecule drugs for MPS II. Conventional small molecule drugs can be taken orally as a pill and have the potential to reach the brain in order to treat the more severe forms of MPS II, unlike enzyme replacement therapy (Elaprase) which can’t cross the “blood-brain barrier”. Our approach is to develop compounds for so-called Enzyme Enhancement Therapy (or EET; aka “Pharmalogical Chaperone Therapy”). This is an approach to treatment that has shown great promise in other lysosomal storage disorders, e.g., Gaucher’s and Fabry disease, but has yet to be tried for the mucopolysaccharidoses such as MPS II. EET works by having a small molecule drug (a “chaperone”) attach itself to the defective enzyme, in this case iduronate sulfatase, and stabilizing it so it can do its intended job: to degrade the mucopolysaccharides in the cell. In order to prepare compounds for EET that are suitable for testing we need to synthesize small molecules that resemble the sugar iduronic acid, the component of the mucopolysaccharides that is degraded by iduronate sulfatase. The first step of this process is to prepare iduronic acid itself and then to make some chemical modifications to it. Iduronic acid is quite a complex sugar and is not commercially available, so in the first year of this project we have focused on developing methods of preparing iduronic acid from cheap and readily available glucose. We have investigated three different methods and one of them has so far shown the most promise. We have been able to prepare an important derivative of iduronic acid with modifications in the desired parts of the molecule. Our next aim is to transform this key intermediate into a range of compounds for testing as chaperones for iduronate sulfatase.
The goal of this project is to study whether the choroid plexus could be made to produce NAGLU-IGF2 into the ventricles of the brain, and whether this will improve brain lysosomal storage in Sanfilippo B syndrome mice. The choroid plexus is responsible for production of cerebrospinal fluid, and if it could be made to produce a therapeutic enzyme, it would serve as a potentially permanent source of that enzyme for the brain. We produced a construct containing the gene encoding NAGLU (the enzyme that is deficient in Sanfilippo B syndrome) fused to IGF2 (insulin-like growth factor 2). Manufactured forms of NAGLU lack mannose 6-phosphorylation, limiting their uptake into cells. To circumvent this problem, we attached IGF2 to NAGLU. IGF2 binds the mannose 6-phosphate receptor so that it can get NAGLU into cells without mannose 6-phosphate. Our studies in cells showed that IGF2 greatly improves intracellular uptake of NAGLU. This project began on July 1, 2011. Year 1 milestones achieved include: 1) re-establishment of the Sanfilippo B mouse colony in our laboratory, 2) production of adeno-associated viral vectors containing NAGLU-IGF2, 3) verification that the vectors produce intact, active NAGLU-IGF2 enzyme and 4) injection of adeno-associated vectors into the brain of normal rats. In year 2, we plan to complete the study the distribution of NAGLU-IGF2 in the brain of normal rats, select an effective dose, and perform a study to evaluate its distribution and effectiveness in Sanfilippo B mice. These experiments will provide proof-of-concept for choroid-plexus directed gene therapy for Sanfilippo B syndrome using NAGLU-IGF2.
Alberto Auricchio (review will be available September, 2012)
Fondazione Telethon, Naples, Italy
Gene therapy of MPS VI
Adriana M. Montaño
Saint Louis University
Role of inflammation in pathogenesis of MPS IVA
Morquio A disease (Mucopolysaccharidosis IVA, MPS IVA) is an autosomal recessive disorder, caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Patients with Morquio A disease have accumulation of the glycosaminoglycans, keratan sulfate and chondroitin-6-sulfate, mainly in bone and cartilage, causing systemic skeletal dysplasia. The broad goals of this research are to characterize the immune profile of Morquio A mouse model and to elucidate the role of cartilage and bone inflammatory reactions in the pathogenesis of Morquio A disease through investigation of secreted inflammatory factors involved in cartilage destruction and bone remodeling.
1. Characterization of the immune profile of the Morquio A mouse model.
Immune response to enzyme replacement therapy (ERT) is the principal limitation in the effectiveness of the treatment. The first step in the characterization of the immune profile of the knock-out Morquio A mouse model is to investigate the immune response after ERT.
We have found that Morquio A mice undergoing ERT have: i) the highest immune humoral response towards the recombinant human GALNS enzyme at 14 weeks of treatment, and ii) the highest cellular response at 16 weeks of treatment. This is consistent with previous observations where age of the mice and length of treatment play a role in the levels of immune response.
2. Characterization of expression profiles of genes associated to the pathogenesis of Morquio A disease.
We compared differences in inflammation profile of cartilage between Morquio A and wild type mice. We have found that there is up-regulation of several genes which play important roles in autophagy and apoptosis. We are in the process of quantifying and comparing these results at various ages in cartilage and bone cells of Morquio A and wild type mice.
Richard Steet, Ph.D.
University of Georgia, Athens, GA
Blockade of cathepsin activity and TGF-beta signaling as a therapeutic approach for LSDs
Investigating the molecular pathogenesis of lysosomal diseases such as the MPS and MPS-related disorders is a promising avenue towards the development of new therapies and can aid our understanding of the mechanisms that contribute to the onset and progression of disease symptoms. Over the past several years, we have taken advantage of a zebrafish ML-II model to investigate the pathogenic mechanisms that underlie the cartilage defects associated with this disease. We have identified and confirmed several target proteins (including cathepsin proteases and matrix metalloproteinases or MMPs) that exhibit increased and sustained activity in ML-II zebrafish embryos and hypothesize that inhibition of this excessive protease activity would result in therapeutic correction of ML-II associated phenotypes. In support of this hypothesis, we have demonstrated that inhibition of cathepsin K by genetic and pharmacological means leads to substantial correction of the cartilage defects in ML-II embryos as well as a surprising reduction in the activity of other proteases. Over the past year, we have focused our efforts on determining whether other proteases such as cathepsin L and MMPs contribute to the disease process. Our results demonstrate that cathepsin L is subject to the same sustained activation in ML-II zebrafish embryos that we previously observed for cathepsin K (Petrey et al, Disease Mechanisms and Models, 5(2) 177-90 (2012)). These results are significant since they point to a common mechanism whereby this class of proteases is abnormally activated from immature forms. We believe this activation arises from the hypersecretion of these enzymes into the extracellular space upon loss of mannose 6-phosphate dependent lysosomal targeting. This hypothesis is supported by 1) the observation that levels of the mannose 6-phosphate recognition marker are greatly reduced or absent on the highly active, mature forms of cathepsins K and L in ML-II embryos and, 2) the direct visualization of cathepsin K exclusively within the extracellular space of developing cartilage of ML-II (but not control) zebrafish. Our near-term goals include an assessment of whether cathepsin L inhibition is capable of producing the same therapeutic benefit in cartilage as we noted with cathepsin K suppression. We have also obtained a specific inhibitor to MMP-13, an enzyme whose activity is increased in ML-II zebrafish. We intend to treat our ML-II model with this inhibitor and determine whether any of the phenotypes can be rescued or improved when this protease activity is decreased. Initial results indicate that this inhibitor can act on zebrafish MMP. Lastly, we have begun to manipulate the TGF-beta signaling pathway in ML-II zebrafish embryos to determine how altered regulation of this pathway relates to the disease phenotypes. Since increased cathepsin and MMP activity, and dysregulation of the TGF-beta signaling pathway are common features of many MPS disorders, we believe our results on ML-II will inform our general understanding of the pathogenesis of other LSDs. Furthermore, our findings indicate that abnormal protease activation (in addition to increased expression) is an important factor that should be considered in assessing the pathogenesis of these diseases. Finally, the demonstration that a reduction in cathepsin activity can provide some therapeutic benefit suggests that further investigation into small molecule protease inhibitors for the treatment of MPS and MPS-related disorders is warranted. We thank the MPS Society for their continued support of this research.
Drs. Patricia Dickson and Stephen Kaler
UCLA Harbor, Los Angeles, CA
Choroid plexus-directed viral gene therapy as a source of cerebrospinal fluid NAGLU-IGF2 for Sanfilippo B syndrome
The goal of this project is to evaluate whether adeno-associated virus (AAV) gene therapy vectors can remodel choroid plexus epithelia to produce N-acetylglucosaminidase (NAGLU) fused to insulin-like growth factor 2 (IGF2). The therapeutic purpose underlying our experiments is to enable secretion of the missing lysosomal enzyme into the ventricles of the brain in a mouse model of Sanfilippo B syndrome. Since choroid plexus epithelia turn over at an extremely slow rate, viral transduction of these cells with a correct version of NAGLU-IGF2 would potentially provide a permanent source of the enzyme in the cerebrospinal fluid for global brain distribution.
We produced a cDNA construct containing the gene that encodes NAGLU (the enzyme deficient in Sanfilippo B syndrome) fused to IGF2. Manufactured forms of NAGLU lack mannose 6-phosphorylation, which limits cellular uptake (Fig. 1). In contrast, NAGLU-IGF2 binds to the mannose 6-phosphate receptor so that delivery is greatly enhanced (Fig. 1). We documented robust expression and NAGLU enzyme activity (1.7 units/mg protein) in HEK293T cells transfected with the NAGLU-IGF2 construct. We next generated a recombinant AAV serotype 5 vector harboring NAGLU-IGF2 (rAAV5.NAGLU-IGF2) under the control of a chicken beta actin promoter and cytomegalovirus enhancer. The rAAV5 serotype is known from our previous work to target choroid plexus epithelia (Donsante et al., 2011), and we confirmed this in wild type neonatal mice (Fig. 2a). We then administered 5×1010 vector genomes of rAAV5.NAGLU-IGF2 to the left lateral brain ventricle of 12 week old Sanfilippo B mice (Naglu-/-). Correct ventricular localization technique was confirmed in a subset of mice by dye injection. NAGLU enzymatic activity in brain sections reached several fold-normal (Fig. 2b) and immunohistochemical evaluation detected NAGLU-IGF2 throughout the brain and into neurons (Fig. 2c).
Our ongoing testing is evaluating the safety and efficacy of this approach to prevent or correct brain pathology and neurological abnormalities in older Sanfilippo B mice. This unique therapeutic approach combines the benefits of M6P-independent endocytosis and viral gene therapy to enable efficient NAGLU-IGF2 distribution throughout central nervous system. Our exciting preliminary results were accepted for presentation at the American Society for Gene and Cell Therapy annual meeting (May 2013, Salt Lake City, UT). The potential impact on clinical practice in the field of LSD is high since, if the proposed aims are successfully achieved, the largest current barriers to health for patients with LSDs will be circumvented. In addition, the principles of gene transfer and CSF protein transport being investigated in this project will potentially be useful for other neurometabolic diseases with global effects on brain.
Prof Vito Ferro
School of Chemistry & Molecular Biosciences, the University of Queensland
Small molecule chaperones for EET for MPS II
Final Report: National MPS Society Research Initiative 2011-2013 (extension to 2014)
The aim of this project is the development of small molecule drugs for MPS II. Conventional small molecule drugs can be taken orally as a pill and have the potential to reach the brain in order to treat the more severe forms of MPS II, unlike enzyme replacement therapy (Elaprase) which can’t cross the “blood-brain barrier”. Our approach is to develop compounds for so-called Enzyme Enhancement Therapy (EET; aka “Pharmacological Chaperone Therapy”). This is an approach to treatment that has shown great promise in other lysosomal storage disorders, e.g., Gaucher’s and Fabry disease, but has yet to be tried for the mucopolysaccharidoses such as MPS II. This is thus the first time that this promising approach has been attempted for MPS II. EET works by having a small molecule drug (a “chaperone”) attach itself to the defective enzyme, in this case iduronate sulfatase, and stabilizing it so it can do its intended job: to degrade the mucopolysaccharides in the cell. In order to prepare compounds for EET that are suitable for testing we need to synthesize small molecules that resemble the sugar iduronic acid, the component of the mucopolysaccharides that is degraded by the enzyme iduronate sulfatase. Two approaches have been explored for this purpose: (i) the synthesis of modified iduronic acid derivatives, and (ii) the synthesis of a compound known to bind to iduronate sulfatase, and derivatives of this compound. The initial stages of this process involved developing chemistries to prepare the required compounds. The first set of 8 test compounds are now in hand and preparations are in progress for their testing using purified enzyme and cell preparations from MPS II patients. The testing will be conducted by our collaborators at the Lysosomal Diseases Research Unit (LDRU) in Adelaide. Our collaborators have established a fluorometric assay for determining the affinity of the compounds for the enzyme. In addition, they have identified MPS II patient mutations that are likely to respond to chaperone therapy. Skin fibroblast cells from these patients will be used to test the compounds for chaperone activity, once Human Ethics Committee approval has been obtained. These studies should demonstrate cellular uptake of chaperone molecules and a reduction of cellular mucopolysaccharide levels, and should identify which mutations respond (best) to chaperone therapy.
We anticipate that the biological testing will identify the most promising candidate chaperones for further optimization and provide proof of concept that this is a viable approach for MPS II treatment. This project continues under funding from an MPS Society Grant (for 2013-2015) with the aim of generating sufficient preliminary data to obtain more significant research funding to accelerate this program towards clinical candidates.
Alberto Auricchio
Fondazione Telethon, Naples, Italy
Gene therapy of MPS VI
Adriana M. Montaño
Saint Louis University
Role of inflammation in pathogenesis of MPS IVA
Morquio A disease (Mucopolysaccharidosis IVA, MPS IVA) is an autosomal recessive disorder, caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Patients with Morquio A disease have accumulation of the glycosaminoglycans, keratan sulfate and chondroitin-6-sulfate, mainly in bone and cartilage, causing systemic skeletal dysplasia. The broad goals of this research were to characterize the immune profile of Morquio A mouse model and to elucidate the role of cartilage and bone inflammatory reactions in the pathogenesis of Morquio A disease through investigation of secreted inflammatory factors involved in cartilage destruction and bone remodeling.
1. Characterization of the immune profile of the Morquio A mouse model.
Immune response to enzyme replacement therapy (ERT) is the principal limitation in the effectiveness of the treatment. The first step in the characterization of the immune profile of the knock-out Morquio A mouse model was to investigate the immune response after ERT.
We have found that Morquio A mice undergoing ERT have: i) the highest immune humoral response towards the recombinant human GALNS enzyme at 14 weeks of treatment, and ii) the highest cellular response at 16 weeks of treatment. This is consistent with previous observations where age of the mice and length of treatment play a role in the levels of immune response.
To ameliorate both humoral and cellular responses in Morquio A mice we are looking at oral tolerance as an alternative. This will to not only decrease the need of administration of immune suppressors to avoid immune responses but also will improve the efficacy of enzyme by avoiding presence of neutralizing antibodies to the enzyme used in ERT.
2. Characterization of expression profiles of genes associated to the pathogenesis of Morquio A disease.
The current study sought to understand if and how inflammation impacts the autophagic pathway in cartilage tissue of a Morquio A mouse model.
We performed expression analysis of over 60 genes using qRT-PCR in Morquio A and wild type mice at 1, 3, 9 and 12 months of age.
We have found that inflammatory, apoptotic and autophagic markers were up- regulated in 12-month-old MKC mice compared to age-matched wild-type controls and compared to younger mice. These findings suggest that inflammatory receptors modulate autophagy and apoptosis in cartilage tissue of Morquio A mice. These data could be used in developing future treatment modalities for bone growth abnormalities in Morquio A patients.
Richard Steet, Ph.D.
University of Georgia, Athens, GA
Blockade of cathepsin activity and TGF-beta signaling as a therapeutic approach for LSDs
We continue to take advantage of our ML-II zebrafish model to explore the relevance of cathepsin and matrix metalloproteases in both the cartilage and cardiac pathogenesis of this disease. Our published work in this area has demonstrated that inhibition of cathepsin K by genetic and pharmacological means leads to substantial correction of the cartilage defects in ML-II embryos as well as an unexpected reduction in the activity of other proteases. Using multiple approaches, we have also shown that this protease is hypersecreted from zebrafish chondrocytes and subject to abnormal proteolytic activation to its mature form.
Over the last year, we have begun to assess the role of other proteases (cathepsin L and MMP13) in the cartilage phenotypes. Using a specific inhibitor to MMP-13 (whose activity is increased 10-12-fold in ML-II zebrafish), we showed that a maximal inhibition of 70% could be achieved in embryo lysates. Despite this level of inhibition, no phenotypic correction of the cartilage phenotype was observed in the embryo, suggesting that this protease is not a central contributor to the developmental defects in this particular tissue. MMP13’s contribution to later stages of disease and to other tissues (such as the cardiac defect) warrants further exploration. More recently, we have begun investigating the pathogenic contribution of cathepsin L and have shown that this protease also appears to undergo abnormal proteolytic activation in ML-II embryos.
Since four homologous cathepsin L genes are now known to exist in zebrafish, we are currently characterizing the expression and localization of their individual transcripts. These analyses, which serve as a prelude to genetically manipulating cathepsin L expression, have revealed that one previously unidentified gene, ctsL1b, is the only isoform with increased transcript levels in the ML-II model. This is important, as our original transcript analyses did not distinguish between the four isoforms. This is also consistent with the finding that morpholino-knock down of the cathepsin L1a isoform did not reduce total cathepsin L activity. In light of these new expression studies, we are continuing to manipulate cathepsin L expression, with cts1b as the main gene targeted. Another goal of the proposed research was to address the relevance of altered TGFbeta signaling in ML-II pathogenesis. Investigation of this pathway and its role in various ML-II phenotypes is underway, and has lead to the identification of several molecular hallmarks for both the cartilage and cardiac defects. We are now positioned to determine whether directly manipulating TGFbeta signaling impacts the phenotypes. The demonstration that a reduction in cathepsin activity can provide some therapeutic benefit suggests that further study into small molecule protease inhibitors for the treatment of MPS and MPS-related disorders is warranted.
We thank the MPS Society for their continued support of this research and remain hopeful that the avenues of research that stem from this work can be translated into therapies for MPS and MPS-related disorders.
2010 Research Grant PDF Download
The National MPS Society awarded $471,000 for research grants in 2010. The funding the Society provides has been and continues to be crucial as we move forward with our mission to find the cures.
We received 45 letters of intent from researchers around the world for the seven grants offered in 2010. After reviewing those letters, our Scientific Advisory Board review committee requested full grant proposals from 14 researchers.
All new grant recipients were awarded $60,000 for the two year grant, with half of the total provided each year. We received $60,000 from the Caterina Marcus Foundation, www.caterinamarcusfoundation.org, to fund a general research grant and $52,000 from Insieme per Gabriel, an ML family foundation in Graglia, Italy, for an ML Partnership Grant. We are honored that both foundations selected the National MPS Society as partners to fund this very important research.
An additional $15,000 will support the work of Brains for Brain. The Society will fund $25,000 to support the Lysosomal Disease Networks NIH grant research goals. The funding is designed for the Neuroimaging Core, which will benefit the four MPS projects. The International MPS Network will announce a grant in September for treatment of CNS in MPS III. The Society has allocated $5,000 for that Partnership Grant.
Mark J. Osborn,
PhD University of Minnesota, Minneapolis, MN
Gene therapy for the central nervous system pathology of MPS I
The lysosome acts as the acidic stomach of the cell and contains multiple enzymes responsible for the breakdown of glycosaminoglycans that are normal constituents of the cell that must be turned over and recycled. A loss of a lysosomal enzyme results in accumulation ofglycosaminoglycans resulting in swelling of the lysosome and loss/altered function of the cell resulting in system wide pathology. MPS I (Hurler syndrome) is caused by a mutation to the IDUA gene causing a loss of the IDUA enzyme that acts as a critical enzyme in the breakdown of glycosaminoglycans. One of the most severely affected organs in patients with Hurler syndrome is the brain that shows widespread pathology resulting in severe mental retardation. The available treatment options for Hurler syndrome do not effectively correct the brain pathology therefore we have proposed to test the ability of a novel protein we have developed to reduce the brain pathology of this disease. Our protein is a hybrid comprised of transferrin and IDUA and is able to cross from the blood into the brain and therefore is able to be delivered in a minimally invasive fashion. We will test this protein by delivering a gene encoding it into MPS mice that have been engineered to mimic the human disease. Our pre-clinical testing will provide proof of concept for pursuing similar studies in humans.
Brett E. Crawford, Ph.D.
Zacharon Pharmaceuticals Inc., La Jolla CA,
Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II
The proposed research project aims to produce a new drug for treating mucopolysaccharidosis (MPS). MPS occurs due to the toxic buildup of cellular carbohydrates (glycosaminoglycans) in cells which lead to serious symptoms ranging from physical deformity, cardiac, joint, andneurological dysfunction. Glycosaminoglycan buildup occurs due to mutations that inactivateenzymes that normally degrade these glycans. Through our previously supported research, we have identified compounds that can alter the synthesis of glycosaminoglycans so that they can becleared from patients with MPS. Our most advanced compounds have demonstrated efficacy in MPS models and are able to enter the central nervous system. These compounds represent a critical starting point for the development of a treatment for the neurological aspect of these diseases. Additionally, due to the mechanism of action (by targeting the biosynthesis of glycosaminoglycans), it is possible that this drug will be effective in multiple classes of MPS.The studies we propose here are aimed at identifying the most promising compound for future clinical development.
Elizabeth F. Neufeld, Ph.D.
UCLA. Los Angeles, CA
Making a minigene suitable for gene therapy for MPS IIIB
MPS IIIB is caused by mutations in the NAGLU gene, causing deficiency of the enzyme alpha-N-acetylglucosaminidase, storage of heparan sulfate and (in brain) of many additional substances. If proved safe, administration of the normal NAGLU gene would be the most effective therapy. Like many other lysosomal storage diseases, MPS IIIB is a candidate for gene therapy, requiring only that the normal gene be introduced into a small number of cells, which would manufacture the enzyme and provide it to neighboring cells, a process known as correction.The gene that is used in gene therapy is not the version found in nature, which is too large to administer to cells or animals. The therapeutic gene is the cDNA version, which is smaller. Ten years ago, we cloned NAGLU cDNA and were disappointed to find that the resulting alpha-Nacetylglucosaminidase was poorly corrective. Nevertheless, this cDNA has been used by four laboratories for gene therapy in MPS IIIB mice. Although all reported therapeutic results, two noted that the results were less than expected. Yet there are plans to use this only partially effective cDNA for clinical trials. Our hypothesis is that segments of DNA taken out of the native NAGLU gene may be important to make an enzyme that will be processed the normal way. This proposal is to make a minigene, which would yield a corrective enzyme and therefore be better suited for gene therapy.
Calogera M. Simonaro PhD., Associate Professor
Mount Sinai School of Medicine, New York, New York
A Novel Approach for the Growth & Expansion of Bone Marrow-Derived Mesenchymal Stem Cells in
Mucopolysaccharidoses Type IV and Other Mucopolysaccharidoses
The overall goal of our research is to develop and evaluate new treatment approaches for two important organ systems in the mucopolysaccharidoses (MPS), the bones and joints. The current project is based on recent work showing that an enzyme, recombinant acid ceramidase (rAC), can be used to maintain and expand a unique population of stem cells from the bone marrow. Bone marrow transplantation (BMT) and related gene therapy procedures have been extensively evaluated in MPS patients and/or animal models, with varying degrees of success. While various factors have influenced this outcome, an important limitation is the very low frequency of stem cells within the bone marrow, leading to very low levels of transplanted cells at the disease sites. Direct injection of these cells into these sites helps, however even here the number of surviving cells is very small. Despite these limitations, clinical improvements have been observed, and there is a general agreement in the field that the approach is beneficial, but needs to be enhanced. In this project we will evaluate whether rAC can be used to improve the outcome of BMT, particularly in the bones and joints. Due to the unavailability of a suitable MPS IV animal model that mimics the severe bone and joint disease seen in patients, we will focus our efforts on the MPS VI rat. We will also study the effect of rAC on the growth and transplantation of cells obtained directly from normal and MPS cartilage. If successful, we believe that this approach could greatly improve the outcome of cell-based transplantation procedures in all of the MPS disorders, including MPS IV, and have general applicability to other genetic disorders as well.
Dr. Katrin Kollmann, PhD Partnership Grant with Insieme per Gabriele
University Medical Center Hamburg-Eppendorf , Hamburg, Germany
Skeletal abnormalities in mucolipidosis II alpha/beta Pathomechanisms and therapeutic strategies
Skeletal abnormalities are common symptoms in mucolipidosis II (ML II) and ML III patients leading to a decline in mobility, stiffness and chronic joint pain. In patients bone cells the transport of multiple lysosomal enzymes to lysosomes is altered impairing the function of bone-forming osteoblasts, bone-resorbing osteoclasts and of chondrocytes of the cartilage resulting in osteoporosis. In this study the expression of proteins and genes will be analyzed in cultured bone cells and chondrocytes of a novel ML II mouse model to understand the mechanisms of osteoporosis and to identify novel targets for therapeutical strategies in this disease. Furthermore, ML II knock-in mice will be treated with inhibitors of bone resorption to reduce the osteoporotic phenotype. These experimental approaches might be of relevance especially for ML III and related lysosomal storage diseases with skeletal abnormalities such as MPS VI.
Dr. Andrea Ballabio Caterina Marcus Foundation Grant
TIGEM (Telethon Institute of Genetics & Medicine)
Naples, Italy
Modulating lysosomal function to treat mucopolysaccharidoses
We recently discovered that a master gene controls the function and biogenesis of organelles called lysosomes, structures inside cells which breaks down materials into compounds which can be used or discarded by the cell, as needed. This gene, named TFEB, activates lysosomal genes, induces lysosomal biogenesis and increases the ability of cell to degrade complex molecules. In this grant, we plan to build on this discovery and test novel therapies in vivo for the treatment of Mucopolysaccharidosis (MPS). The possibility of achieving global control of lysosomal function, if successful, would represent a paradigm shift in biology and have enormous implications on the therapy of several lysosomal storage disorders, including MPS.
Dr. Alisdair B. Boraston, PhD
University of Victoria, Victoria, BC, Canada
Discovery and assessment of inhibitor-based chemical chaperones as potential
agents for the treatment of mucopolysaccharidosis IIIB.
The mucopolysaccharidoses are a group of devastating genetic diseases for which there are currently no cures or even effective treatments. Mucopolysaccharidosis IIIB (MPS IIIB), or Sanfilippo syndrome, is one of these diseases that usually results in death by early adulthood. Our ability to study the cause of MPS IIIB at the atomic level will allow us to develop new medicines to treat MPS IIIB and improve the lives of people suffering from this disease.
MPS II
Brett E. Crawford, Ph.D.
Zacharon Pharmaceuticals Inc., La Jolla CA,
“Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II”
With the support from the National MPS Society, we have made significant progress toward developing a new therapeutic approach designed to treat both the neurological and non‐neurological symptoms of MPS. This approach is based on chemical compounds that modify glycosaminoglycan synthesis so that certain lysosomal enzymes are not required to degrade them. The following is a brief description of our progress over the last year:
Improve the Potency of the Lead Compounds. The first goal of our proposed research is to improve the potency of the inhibitors to a level needed for robust efficacy in experimental models and in patients. This is a very important stage in drug development to ensure that the drug can be administered at effective doses and is sufficiently selective to be safely used in humans. Using the cellular assay of MPS that we developed though our previous MPS Society funding, we have successfully increased the potency of the compound by over 100-fold.
Identify Safe and Effective Drug Candidates. With significant progress on the potency, we are now focused on improving other drug like properties required for clinical development. These features include the selectivity, pharmacokinetics, stability, and formulation. Through these studies we aim to identify a potent analog that has all of the characteristics in a drug candidate that is suitable for clinical studies in MPS patients.
Through the next year of support, we are excited to test these optimized compounds in the mouse models of MPS. Due to the broad efficacy of this therapeutic approach we expect to test these compounds in models of MPS I, II, and III.
Partnership for Further Development. Based on the progress that we have made through MPS Society grants, NIH grants, and private investors, we are also happy to report that we have entered into a strategic research collaboration with Pfizer to jointly develop these compounds as novel therapeutics for the treatment of MPS.
MPS I
Mark J. Osborn, PhD (eight month extension awarded)
University of Minnesota, Minneapolis, MN
Gene therapy for the central nervous system pathology of MPS I
MPS II
Brett E. Crawford, Ph.D.
Zacharon Pharmaceuticals Inc., La Jolla CA
Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II
With the continued support from the National MPS Society, the National Institute of Neurological Disorder and Stroke and Pfizer, we have made significant progress toward developing a new therapeutic approach designed to treat both the neurological and non-neurological symptoms of MPS. This approach is based on compounds that modify glycosaminoglycan synthesis so that certain lysosomal enzymes are not required to degrade them.
The funding from the National MPS Society has provided critical support that has helped us progress this drug development from an idea to an NIH grant supported program and recently to a partnership with Pfizer. The following is a brief description of our progress over the last year:
Testing the In Vivo Efficacy of Lead Compounds. Through the first year of support, we identified a series of analogs with improved potency in cellular models of MPS. Our most potent analogs are active in the 100 to 500 nM range, a 100-fold improvement from the original compound. Over the last year, these compounds have been evaluated for their drug-like properties and pharmacological characteristics in a wide range of enzymatic, cellular, and rodent models. Three of these potent compounds with acceptable pharmacological properties were recently tested in the MPS IIIA mouse model. These in vivo studies demonstrated that all three compounds reduced the lysosomal accumulation of glycosaminoglycans n the brain. Future studies will explore the minimal effective dose and dose required to achieve a phenotypic benefit in the mouse model.
Expanded Drug Discover Effort. With the additional support of Pfizer over the last year, we have also expanded our efforts to identify additional therapeutic approaches to MPS. We have used our cellular model of MPS and the Sensi-Pro® assay to screen hundreds of compounds from Pfizer’s collections that are potent inhibitors of known drug targets. Our goal is to identify new drug targets that could accelerate the clinical testing of active agents in MPS patients. These studies have revealed a number of potentially active compounds that we are currently characterizing further.
In the nest year we will continue to optimize the drug-like properties of the most potent inhibitors, explore novel approaches to MPS, and test the improved compounds in the MPS mouse models. We are optimistic that a novel therapy will emerge from these studies and move closer to clinical testing in the near future. We sincerely appreciate the dire need for effective therapies for these devastating diseases and are committed to bring a new therapy to the clinic as soon as possible.
Elizabeth F. Neufeld, Ph.D.
David Geffen School of Medicine at UCLA, Los Angeles, CA
Making a minigene suitable for gene therapy for MPS IIIB
Calogera M. Simonaro, PhD
Mount Sinai School of Medicine, New York, NY
A Novel Approach for the Growth & Expansion of Bone Marrow-Derived Mesenchymal Stem Cells in
Mucopolysaccharidosis Type IV and Other Mucopolysaccharidoses
The overall goal of this research project was to evaluate the use of a recombinant enzyme (acid ceramidase, rAC) produced and studied in our laboratory to improve the outcome of cell-based therapies for the MPS diseases. An important and debilitating feature of MPS is progressive cartilage destruction leading to the development of severe arthritic joint disease. At the present time there are no suitable methods to prevent these abnormalities in MPS, and current enzyme replacement (ERT) and bone marrow transplantation (BMT) therapies have limited effects. We have previously found that glycosaminoglycan (GAG) storage in MPS activates numerous inflammatory and other signaling pathways, leading to cartilage cell (i.e., chondrocyte) death and cartilage destruction. Among the many changes in MPS, there is an elevation of the pro-death fat or lipid called ceramide. Based on this we proposed that rAC could be used to improve the survival and integrity of MPS chondrocytes, both in the laboratory (cell culture) and in animal models of MPS. rAC is the enzyme that degrades the pro-death lipid ceramide, providing a basis for this hypothesis. During the course of this project we determined that the addition of rAC to normal animal (human, rat, horse etc) chondrocytes maintained in the laboratory improved their growth and quality, as determined by several established methods to assess cartilage integrity (e.g., expression of cartilage-specific collagen etc). Moreover, we found that these effects were even more pronounced using chondrocytes obtained from several animal models of MPS. Due to the GAG accumulation in MPS cells and subsequent downstream changes, these cells grow very poorly and lose their cartilage-like properties, even more than normal cells. In the presence of rAC, these features were significantly improved. We also tested the effects of rAC on the growth and properties of stem cells obtained from the bone marrow of MPS animals (i.e., bone marrow mesenchymal stem cells, MSC). We found that addition of rAC to normal and MPS bone marrow cells increased the production of MSC about 2-fold, and also significantly improved their ability to become chondrocytes. We tested this using bone marrow from several MPS animal models, and found similar results. We also obtained bone marrow from mice expressing a protein called green fluorescence protein (GFP), and also found similar results. Based on these observations we have begun to evaluate whether cells (MSC or chondrocytes exposed to rAC) grow better after they are transplanted into MPS animals, and whether they have an enhanced effect on repairing or preserving the cartilage disease. These studies have been initiated and are ongoing. Thus, our ongoing goal continues to be to develop improved methods to repair the defective cartilage in MPS patients. Funds from this grant have provided essential information that has moved us closer to this goal.
Katrin Kollmann, PhD (Partnership grant with Insieme per Gabriel)
University Medical Center Hamburg-Eppendorf , Hamburg, Germany
Skeletal abnormalities in mucolipidosis II alpha/beta – Pathomechanisms and therapeutic strategies
The lysosomal storage disease mucolipidosis type II (MLII) is caused by defects in the GlcNAc-1-phosphotransferase. The phosphotransferase is an enzyme complex composed of six subunits (a2b2g2) that catalyzes the first step in the formation of the mannose 6-phosphate (M6P) recognition markers on lysosomal hydrolases. The M6P recognition marker is important for the efficient transport of newly synthesized lysosomal proteins/hydrolases to lysosomes. In MLII with mutations in the gene encoding the alpha/beta subunits of the complex (MLII alpha/beta), lysosomal hydrolases are not modified with M6P residues, and therefore many lysosomal hydrolases are missorted and do not reach lysosomes. The deficiency of hydrolases in the lysosomes leads to lysosomal dysfunction and the accumulation of undegraded material in different cell types of the body. Severe skeletal abnormalities accompanied by a decline in mobility and chronic joint pain are features of MLII alpha/beta. We generated a mouse model for MLII alpha/beta by the insertion of a mutation into the murine Gnptab gene (c.3082insC) that is homologous to the mutation GNPTAB c.3145insC detected in MLII patients. The MLII mice show all characteristic biochemical alterations and clinical features found in the human MLII disease and allow the analysis of underlying pathogenetic cellular mechanisms. MLII mice show an increased lethality, reduced mean body weight and body length, and display skeletal alterations like abnormal spine curvature and osteoporosis1. We investigated the bone pathology in detail by biochemical, histomorphometric, histochemical and immunological methods to characterize alterations and identify pathomechanisms affecting the bone metabolism. Electron microscopic analyses demonstrated the formation of storage lysosomes in osteocytes and osteblasts but not in osteoclasts. To analyze the targeting defect in the bone we cultured primary cells like osteoblasts and osteoclasts of wildtype and the MLII mice and determined the steady state expression level, sorting, proteolytic processing and the half-live of several enzymes such as tartrate resistant acid phosphatase (TRAP), cathepsin D, Z and K. Pulse-chase and real-time PCR experiments on cultured fibroblasts, osteoblasts and osteoclasts indicated that the rate of synthesis is similar in MLIIcells compared to wildtype cells whereas the sorting efficiency to lysosomes was affected resulting in their hypersecretion into the medium. The extent of missorting, however, depends on the lysosomal enzyme examined. Thus, β-hexosaminidase and TRAP, were found to be highly reduced in MLIIfibroblasts, osteoclasts and osteoblasts whereas the steady state expression level of proteins like cathepsin D were unchanged. Our data indicate that subpopulations of lysosomal hydrolases appear to be more affected by the loss of M6P residues than others transported to lysosomes via M6P-independent pathways. Whereas in osteoclasts the missorting of lysosomal proteins lead to an increased bone resorption capacity in vitrothe consequences of their mistargeting in osteoblasts are unclear. Microarray analyses carried out in cultured osteoblasts and osteoclasts from wildtype and MLII mice revealed changes in the expression of several genes which have to be confirmed by independent methods. Current studies are focussed on i) the isolation and identification of osteoblast-specific M6P-containing proteins that are directly involved in the regulation of bone remodelling, and ii) the pharmacological intervention of altered bone metabolism in MLII.
1-Kollmann K, Damme M, … (2012) Lysosomal Dysfunction Cause Neurodegeneration in Mucolipidosis II “Knock-in” Mice. Brain, in press
This work was presented on the ESGLD (European Study Group on Lysosomal Diseases) workshop in Helsinki 2011, where it was selected for oral presentation. At the annual conference of the APS 2012 (Working group for paediatric metabolic disorders in the german society for children medicine) it was awarded the poster prize.
Dr. Andrea Ballabio (Partnership grant with Caterina Marcus Foundation)
TIGEM, Naples, Italy
Modulating lysosomal function to treat MPS
A. SPECIFIC AIMS
Aim 1: Development of tools for in vivo TFEB activation
Aim 2: Evaluation of the therapeutic effects of in vivo TFEB overexpression in MPSIIIA mice.
To study the effects of TFEB overexpression in vivo in both wild-type mice and in the mouse models of MPSIIIA, we generated a conditional gain-of-function (TFEB-COND-GAIN)mouse line. Time-and/or tissue-specific expression of the transgene can be obtained by crossing the transgenic mouse line with a strain carrying the CRE recombinase. As a first test of the system we generated two founder lines specifically overexpressing Tcfeb in the liver using the Albumin-CRE strain that expresses CRE in the hepatocytes. These lines show different levels of TFEB overexpression. One overexpresses TFEB approximately 3-fold normal levels, while the other approximately 20-fold. We observed that TFEB overexpression in liver resulted in the activation of TFEB target genes. To generate brain specific expression of TFEB, the TFEB-COND-GAIN mice were crossed with a NESTIN-CRE strain, that expresses CRE in the brain and central nervous system.
Unfortunately, we observed that the first line tested showed embryonic lethality. We believe this could be due a wider expression pattern of NESTIN-CRE in the developing embryo, and by the high levels of TFEB overexpression. We are repeating the experiments with the founder line that shows lower expression levels of TFEB and by using a different brain specific CRE line, GFAP-CRE that has a more restricted expression pattern. In the meantime, we had obtained very encouraging results on the function of TFEB overexpression in cellular models of lysosomal storage disorders (LSDs), both MPSIIIA and multiple sulfatase deficiency (MSD). We had evidence that TFEB overexpression increased lysosomal exocytosis in cultured HeLa cells, we tested whether we observed the same effect in mouse embryonic fibroblasts derived from the murine models of MSD and MPSIIIA. TFEB overexpression in these cells types resulted in a significant increase of LAMP1 on the plasma membrane and of lysosomal enzymes into the culture medium, hallmarks of lysosomal exocytosis. This indicates that LSD cells efficiently respond to TFEB-mediated induction of lysosomal exocytosis.
Therefore, we evaluated the effect of TFEB overexpression on the clearance of GAGs in glia differentiated neuronal stem cells (NSCs) isolated from mouse models of MSD and MPSIIIA. TFEB overexpression resulted in a striking reduction of alcian blue-stained GAGs in both MSD and MPSIIIA NSC-derived glial cells (Figure 1A). The latter result was further confirmed by pulse-and-chase experiments using H3 glucosamine to label GAGs, showing a significant reduction of the levels of labeled GAGs after 48 hr of chase in both MSD and MPSIIIA NSC-derived glial cells overexpressing TFEB (Figure 1B). Finally, EM analysis revealed that TFEB-mediated clearance of GAGs in TFEB-overexpressing MSD and MPS-IIIA cells was associated with both significant reduction of cellular vacuolization and recovery of normal cellular morphology (Figure 1C). These results indicate that TFEB overexpression results in an increased lysosomal exocytosis that leads to increased cellular clearance. We have completed the generation of an adeno-associated type 2/9 virus (AAV2/9) that carries TFEB-3xflag under the control of a strong TBG promoter. As a first pilot study we injected this vector systemically into adult multiple sulfatase deficiency (MSD) mice. This mouse model allowed us to treat the mice with a systemic injection, that is technically less challenging than direct intra-cerebral injections.
To this end, we injected systemically AAV2/9-TFEB-3xflag into adult MSD mice. One month after injection, several tissues were collected to monitor transduction efficiency and GAG storage. AAV-mediated TFEB delivery resulted in efficient TFEB transduction and significant reduction of GAG staining in liver and skeletal muscles, as detected by alcian blue staining and GAG quantification (Figure 2A,B). Subsequently, we investigated whether TFEB-mediated clearance of GAGs resulted in the reduction of the pathologic hallmarks of MSD, such as macrophage infiltration and apoptosis. We found a striking reduction of CD68-positive cells in AAV-TFEB injected MSD mice compared with untreated mice (Figure 2C). Most importantly, we also observed a significant reduction of TUNEL-positive cells (Figure 2D) (Medina el at Development Cell Volume 21, Issue 3, 421-430). These results indicate that TFEB activation of lysosomal exocytosis reduced both primary accumulation of GAGs and secondary pathological processes associated with LSDs such as inflammation and cell death. Our next challenge is to treat MPSIIIA mice with this vector in the brain directly.
Dr. Alisdair B. Boraston
Department of Biochemistry and Microbiology, University of VictoriamVictoria, Canada
Discovery and assessment of inhibitor-based chemical chaperones as potential agents for the treatment of mucopolysaccharidosis IIIB.
HYPOTHESIS – Mutant forms of human NAGLU, which cause the MPS IIIB phenotype, are destabilized by the mutations, but not rendered non-functional, and can be chaperoned to the lysosome by specific inhibitors of a-N-acetylglucosaminidase (NAGLU) inhibitors (chemical chaperones) to result in elevated levels of lysosomal NAGLU activity.
GENERAL APPROACH – 1) Generation of potent and selective inhibitors of NAGLU. We are combining synthetic chemistry and X-ray crystallographic analysis of a model protein in complex with synthesized inhibitors to generate compounds that are selective for NAGLU. 2) Candidate inhibitors are being assessed in a Chinese Hamster Ovary (CHO) cell model of MPSIIIB. The readout in this model assay is increased NAGLU concentration and activity in lysosomes upon treatment with our compounds.
BACKGROUND RESULTS – Prior to receiving funding from NMPSS we established using biochemical, structural, and cellular assays that inhibitory compounds based on piperidine and indolizine scaffolds could be made and would function as reasonably effective and selective chemical chaperones in our cellular assay, providing the basis for this work. Our endeavours were to focus on expanding the number of compounds that display the required properties for chaperones namely affinity, solubility, bioavailability and selectivity as well as expanding the model system to include other naglu mutations.
PROGRESS – Towards investigating inhibitor scaffolds using synthetic methodologies that potentially could result in compounds that are potent inhibitors of NAGLU, we have prepared a large number of compounds and are at different stages of their evaluation as inhibitors of NAGLU. Efforts to date have centered on the use of scaffolds demonstrated to be important in inhibiting glycosidases in general. Due to the synthetic difficulties that have resulted in some of the scaffolds syntheses, during the study we decided to first focus on hydroximolactone and piperidine scaffolds (see above figure) as these could be prepared in a more robust fashion.
We have completed the synthesis of a library of compounds for these scaffolds and are at different stages of evaluating them as inhibitors of NAGLU. Initial results have shown whilst they are modestly potent against NAGLU, these studies and the evaluation of roughly 20 crystal structures of bacterial NAGLU in complex with these inhibitors reveal them to lack selectivity for the enzyme over a functionally related human enzyme. This information, although disappointing, will guide us in the future preparation of selective compounds for NAGLU.
Despite the compounds above not being selective for NAGLU, we have been assessing the chemical chaperone potential of them in a CHO cell model of MPSIIIB that incorporates a single known mutation of naglu. We have assayed the most potent compounds found to date as chemical chaperones in the model against the six mutants that we had prior to receiving funding from NMPSS. Nine new mutants have been prepared in this study, bringing the total to fifteen mutants in our library. Again it was disappointing to find that of the compounds that have been assayed, even though they were not toxic at high concentrations, they were not able to increase the levels of mutant NAGLU activity above control levels with any of our mutants. The nine new mutants were also not active against our initial piperidine and indolizine compounds that we had prior to receiving funding from NMPSS. We are now assessing the observed weaker binding compounds of NAGLU, as well we will assess the compounds that are yet to be evaluated as inhibitors of NAGLU, as chemical chaperones in future work. These results have also guided us in the future development of compounds that display the required properties for chaperones namely affinity, solubility and bioavailability.
2009 Research Grants
2009 Research Grant PDF Download
All grant recipients were awarded $80,000 for the two year grant, with half of the total provided each year. Dr. Cosma received the MPS II grant, Drs. Esko and Fraldi received the MPS III grants, and Drs. Ponder and Simonaro received the general MPS research grants.
An additional $7,000 for mucolipidosis research will be provided as a partnership grant to ISMRD (International Society of Mannosidosis and Related Diseases). In support of the Lysosomal Disease Networks NIH grant research goals, the Society will fund $25,000 for the Neuroimaging Core which will benefit the four MPS projects.
Dr. Maria Pia Cosma
TIGEM, Naples, Italy
AAV2/5CMV-IDS therapy in MPSII mice: correction of CNS defects through IDS delivery across the blood-brain barrier.
Children affected by mucopolysaccharidosis type II (MPSII; Hunter syndrome) lack the activity of the enzyme iduronate 2-sulfatase (IDS). They accumulate compounds in their body that gradually kill their cells and damage all of their visceral organs. A gene therapy approach was initiated to treat this central nervous system (CNS) disease in a mouse model of MPSII. Affected pups were injected with viral particles that targeted all of the visceral tissues. High levels of active IDS were produced, secreted into the plasma and also taken up by the brain. This approach gave important results, as the mice were cured of their visceral organ defects, and surprisingly, they also showed amelioration of the CNS phenotype. We now plan to extend this approach to adult and juvenile MPSII mice and to more specifically study how the IDS enzyme reaches the brain, in terms of its crossing of the blood-brain barrier, which was thought not to be permeable to high molecular weight proteins, such as the IDS. We plan to carry out these studies with a variety of different approaches. If successful, our studies should allow us to set up more efficient treatments for the cure of the CNS phenotype of patients with Hunter syndrome.
Dr. Jeffrey Esko
University of California, San Diego, CA
Substrate reduction strategy for MPS IIIA
Mucopolysaccharidoses (MPS) are inherited metabolic disorders in which cellular polysaccharides (glycosaminoglycans) can no longer be degraded, causing aberrant storage of partially degraded material in lysosomes. Children born with these diseases exhibit developmental abnormalities, organ failure and mental retardation, defects that often result in death within the first few decades of life. A subset of MPS diseases result from enzyme deficiencies required by cells to degrade a class of glycosaminoglycans known as heparan sulfate. This research proposal will test if altering heparan sulfate biosynthesis is an effective method of preventing its accumulation in one of these diseases, specifically MPSIIIA. The approach consists of genetically disrupting heparan sulfate biosynthesis in MPSIIIA patient cell lines and mouse models. Its efficacy will be assayed by reduction of lysosomal storage and restoration of normal cellular turnover of glycosaminoglycans. Positive results would justify and encourage the development of small molecule inhibitors of heparan sulfate biosynthesis as a way to accomplish substrate reduction therapy in patients. The major advantage of substrate reduction is that these agents might access the brain where glycosaminoglycan storage is highly detrimental and existing therapies appear ineffective
Dr. Alessandro Fraldi
TIGEM, Naples, Italy
Developing a systemic AAV-mediated gene therapy to cross the blood-brain barrier and treat the brain pathology in MPS IIIA
Mucopolysaccharidosis type IIIA (MPS-IIIA) is an inherited disease caused by the deficiency of sulfamidase (SGSH), a gene that encode an enzyme needed for the degradation of a large macromolecule called heparan sulfate. As consequence, such substrate accumulates in the cells and tissues of the affected patients causing cell damage. The central nervous system is the predominant target of damage and in fact, the MPS-IIIA patients experience severe mental retardation and neuropathological decline that ultimately leads to death. Gene therapy is a therapeutic option for several inherited diseases. The aim of gene therapy is to substitute the defective gene with a functional one. Often a modified not-pathogenic virus is used as vehicle to transport the gene in the affected tissues. In this study we will test the efficacy of a therapeutic approach based on the delivery, via intravenous injection, of an adeno-associated virus (AAV) bearing a functional SGSH. The AAV have a tropism for the liver, so that upon injection the virus will reach the liver that consequently will produce the functional SGSH. The functional SGSH will be then secreted from the liver and will enter into the brain throughout the blood torrent. Importantly, the SGSH will be opportunely modified to be secreted more efficiently from the liver and to make it able to efficiently pass the blood-brain barrier and transduce the brain.
Dr. Katherine Ponder
Washington University, St. Louis, MO
The role of cathepsin K in cardiac valve disease in MPS
Mucopolysaccharidosis (MPS) is due to a genetic deficiency in the activity of an enzyme that degrades glycosaminoglycans. One of the serious manifestations of MPS is the development of heart disease, which can result in reduced delivery of oxygenated blood to the body and require surgery to replace the valve. This can involve thickened heart valves that block the flow of blood into the heart. Heart valves can also be leaky, which allows blood to flow in the wrong direction. The goal of this project is to understand what causes heart valves problems, and to identify a therapy to prevent these heart valve abnormalities from developing. Collagen is the major protein that provides strength to the heart valves. We have found that the amount of collagen is markedly reduced in heart valves of MPS I and MPS VII dogs, and propose that this is what weakens the valve. We hypothesize that reduced amounts of collagen are due to abnormally high levels of an enzyme that can degrade collagen, cathepsin K. It that is the case, it might be possible to prevent heart valve disease with inhibitors of cathepsin K that are currently being used to treat osteoporosis. This project may identify a drug to prevent the development of heart valve disease in MPS.
Dr. Calogera Simonaro
Mount Sinai School of Medicine, New York, NY
Novel anti-inflammatory therapies for the mucopolysaccharidoses
Enzyme replacement therapy (ERT) is currently available for three MPS diseases, although the effects of this therapy on bone and cartilage are very limited. Thus, new treatment approaches are clearly needed, alone or as adjuncts to ERT. Our research will use animal models to explore such new therapies. In particular, we will comprehensively evaluate the bones and joints of MPS VI animals treated with the FDA-approved anti-inflammatory drug, Remicade. This drug targets the inflammatory pathway we have found to be activated in MPS patients (TLR4). Our results to date have shown that Remicade can substantially reverse or prevent inflammation in MPS VI rats, and we now plan to comprehensively evaluate the bones and joints in animals treated with Remicade alone or in conjunction with ERT (Naglazyme). Since Remicade is currently FDA-approved for the treatment of arthritis and other inflammatory diseases, we are hopeful that completion of these animal studies will lead to clinical trials and approval for MPS patients. We will also complete the analysis of an important proof of principle experiment in which the TLR4 inflammatory pathway is inactivated in MPS VII mice. These results will provide the basis for the continued development of anti-inflammatory treatment strategies for MPS VI and other MPS disorders, and identify new molecular targets for drug therapy.
Dr. Sara Cathey One year Partnership Grant with ISMRD
Greenwood Genetic Center, North Charleston, SC
Natural history study for mucolipidosis
In 2008 and 2009 Partnership Grants with ISMRD supported natural history studies of Mucolipidosis (ML) II and ML III. The ML Project of the Greenwood Genetic Center (GGC) is an ongoing effort to understand the disease course in people affected by these rare conditions. Fourteen individuals with ML II or III and their families participated in GGCs Second ML Clinic, held July 27-29, 2009 at the South Carolina Center for the Treatment of Genetic Disorders in Greenwood, South Carolina. During this special clinic, participants were evaluated by clinical geneticists, psychologists, an orthopedic surgeon, and an ophthalmologist. All patients had skeletal x-rays to evaluate the progression of bone disease. Many patients (and their parents!) provided samples for laboratory studies. Most of these same patients participated in the first ML Clinic held in 2006 at GGC. ML affects people from around the world, and each patient can contribute to better understanding. The natural history study went international in November 2009 when ML patients from Australia and New Zealand participated in special clinics held in Sydney, Australia and Wellington, New Zealand.
The ML Project has had a positive impact in many ways, including important contributions to the medical literature about ML. Large collections of clinical and laboratory data have been established. Samples collected are used by scientists at GGC and researchers around the world. Patients and their doctors have another source of information and support. Following a group of patients with this rare disease over time lets us learn the natural history of the illness, evaluate the usefulness of potential therapies, and set sights on effective treatments. To conquer the illness we have to learn about the people affected by ML.
Lysosomal Disease Network, one year grant
University of Minnesota, Minneapolis, MN
Neuroimaging Core activities of the projects of the LDN grant during the first year of work
Progress Report for MPS Society Award This award is being used to support the Neuroimaging Core of the Lysosomal Disease Network, specifically the work of Dr. Alia Ahmed in volumetric neuroimaging (about 50% of her salary). The award thus supports the following LDN studies, all of whom employ neuroimaging:
The neuroimaging core has four functions, 1) to collect imaging data in a comparable manner suitable for natural history determination, 2) Develop and validate disease specific standardized imaging protocols, semi-quantitative MRI clinical ratings scales, and formal quantitative measures to be used as biomarkers for natural history and clinical trials, 3) demonstrate that standardized protocols will identify optimal imaging sequences, appropriate intervals for follow-up, and best clinical practices, 4) archives of scans for educational purposes. Dr. Ahmed’s work supports items 2 and 3. With regard to optimal imaging seqences, protocols have been established by our neuroimaging core for volumetric analysis.
For quantitative volumetric analysis of scans, we have established methods for reliable determination of hippocampus and amygdala using a three dimensional manual tracing program called BRAINS2. Dr. Alia Ahmed is expert at this demanding task. Prior to funding, to develop her skills, she became proficient in tracing on 10 hippocampi of tremor patients and 5 caudates of normal subjects, using ImageJ, a two dimensional program. We decided to change to BRAINS2, which is a three dimensional program allowing tracing on coronal, saggital, and axial images which can be then combined to produce a structural image. To trace the hippocampus for each patient, she traced one image in a saggittal section and two in a coronal section, one with and one without white matter. Thus for each brain scan she traces each (left and right) hippocampus 3 times, making 6 tracings.
For the amygdala, which also requires manual tracing, for each brain scan she does one sagital and one coronal tracing of the left and of the right amygdala. So far to trace the hippocampus, she has traced 42 MPS I brain scans, 9 MPS II, 10 MPS III, 7 MPS VI, 2 MPS VII , and 9 control brain scans ( Total 79 brains scans X 6 tracings = 474). For the amygdala, she has traced 16 brains (we are only doing it for MPS II and III) totaling 64 tracings. She has also traced the caudate in 13 patients totaling 26 tracings. Thus, in summary, Dr. Ahmed has successfully traced hippocampus on 70 brains of MPS children and 9 controls and has successfully traced the amygdala in 16 patients. These tracings can be seen in three dimensional space and we are anticipating that we can evaluate changes in configuration as well as volume over time. We are comparing some of Dr. Ahmed’s tracings to an automated program called FreeSurfer which is implemented at the Minnesota Supercomputer Center (they have provided a grant to cover our costs). She also did intra rater reliability studies on ten brain scans (rating each one twice) and now is working on inter rater reliability studies (comparing her ratings with those of Dr. Nestrasil, our expert in image acquisition and other methods of image analysis.
Thus far, excellent manual tracing reliabilities on MPS I patients and preliminary data on brain volumes in 18 MPS I patients and 9 MPS II patients have been obtained. The results of the reliability study were presented at the WORLD symposium in February; this presentation is now being written for publication. It is attached to this progress report as well as the volumetric comparisons of attenuated MPS I and MPS II. In addition, clinical ratings scales (such as the Matheus scale for MPS disorders) are now being refined to increase their reliability. Dr. Ahmed is also carrying out this analysis together with a member of our radiology department to determine its reliability and to determine if we can improve its sensitivity. With respect to her involvement in the four studies: she has analyzed brains for 40 children in the Shapiro study, 25 for the Polgreen study (overlaps with Shapiro study), 10 for the Potegal study, and 3 for the Dickson study. Funds have been used for 1) Dr. Ahmed’s salary, 2) a computer for her image analysis, and 3) travel for meetings and training.
Abstract:
Ahmed, A., Nestrasil, I., Rudser, K,, Shapiro, E. Reliability of manual and automated tracing of hippocampal volumes in MPS patients and normal controls: A report of the neuroimaging core of the lysosomal disease network. Molecular Genetics and Metabolism, 99 (2); 2010, S9.
Dr. Maria Pia Cosma
TIGEM, Naples, Italy
AAV2/5CMV-IDS therapy in MPSII mice: correction of CNS defects through IDS delivery across the blood-brain barrier.
Results funded by the MPS society have been published in the following paper:
Polito V and Cosma MP (2009). IDS crossing of the blood-brain barrier corrects CNS defects in MPSII mice. AJHG 85(2):296-301.
Abstract
Mucopolysaccharidosis type II (MPSII), or Hunter syndrome, arises from a deficiency in iduronate 2-sulfatase (IDS), and it is characterized by progressive somatic and neurological involvement. The MPSII mouse model reproduces the features of MPSII patients. Systemic administration of the AAV2/5CMV-hIDS vector in MPSII mouse pups results in the full correction of glycosaminoglycan (GAG) accumulation in visceral organs and in the rescue of the defects and GAG accumulation in the central nervous system (CNS). Remarkably, in treated MPSII animals, this CNS correction arises from the crossing of the blood-brain barrier by the IDS enzyme itself, not from the brain transduction. Thus, we show here that early treatment of MPSII mice with one systemic injection of AAV2/5CMV-hIDS results in prolonged and high levels of circulating IDS that can efficiently and simultaneously rescue both visceral and CNS defects for up to 18 months after therapy. We are now testing if the treatment of the CNS defects can be also achieved in juvenile and adult MPSII mice. For that we injected groups of ids-/y animals with AAV2/5CMV-IDS viral particles. We are evaluating the IDS activities in the plasma and GAG contents in the urines to monitor the efficiency of the therapy.
In addition the mice were tested with rotarod tests which evaluate the sensorimotor coordination. Indeed, we have truly characterized groups of ids-/y mice at different ages and found that Purkynje cells in the cerebellum strongly degenerate through the progression of the disease. Thus we are testing the rescue of the cerebellum defects after the therapy. In addition to these gene therapy experiments we are also setting up enzyme replacement protocols to treat simultaneously the visceral defects and the CNS features of the MPSII mice.
We are developing efficient protocols that allow the IDS to cross the blood brain barrier, to correct CNS phenotype and to reverse neurobehavioural features.
Dr. Jeffrey Esko
University of California, San Diego, CA
Substrate reduction strategy for MPS IIIA
The original focus of this grant was to demonstrate that substrate reduction would prove effective as a treatment for Sanfiippo syndromes. We found that reducing the level of reduced heparan sulfate biosynthesis in MPSIIIa fibroblasts by ~40% diminished lysosomal storage by ~50%. These studies are limited to short-term experiments because of the way that we silenced the expression of certain genes in the system. To get at this question in vivo, we have crossed the MPSIIIa mouse with mice carrying mutations in genes involved in heparan sulfate biosynthesis. The objective is to test the impact of substrate reduction on lysosomal storage and pathology in specific cell types in vivo, specifically hepatocytes, astrocytes, neurons and macrophages. We should have our first results in the next 6 months.
We also found that MPSIIIa cells (as well as MPSIIIb, MPSIIIc, and MPSIIId cells) store a significant amount of chondroitin/dermatan sulfate in addition to heparan sulfate. This finding was unexpected since the primary genetic defects in Sanfilippo syndrome are in enzymes specifically involved in heparan sulfate biosynthesis. Enzyme replacement therapy using recombinant sulfamidase, the enzyme missing in MPSIIIa, resolved both the primary and secondary storage (Fig. 1), suggesting that the accumulation of heparan sulfate caused the secondary storage of chondroitin/dermatan sulfate.

To understand the cause of secondary storage, we assessed whether heparan sulfate inhibited enzymes involved in chondroitin/dermatan sulfate degradation. We found that heparin and heparan sulfate strongly inhibited the enzyme iduronate-2-sulfatase (IC50 values of ~3 ?g/mL and ~14 ?g/mL, respectively). We are currently testing whether supplementing iduronate-2-sulfatase can reduce secondary storage and whether strategies that reduce both primary and secondary substrate accumulation are more effective at treating lysosomal storage.

Dr. Alessandro Fraldi
TIGEM, Naples, Italy
Developing a systemic AAV-mediated gene therapy to cross the blood-brain barrier and treat the brain pathology in MPS IIIA
The aim of this project is to develop a low-invasive systemic gene therapy strategy based on the intravenous injection of AAV serotype 8. This serotype displays high tropism to the liver and will be used to delivery of an engineered gene encoding a chimeric modified sulfamidase optimized to be: (i) highly secreted from the liver thus reaching high levels of circulatingenzyme in the blood stream; (ii) to efficiently cross the BBB.
Construction and validation of the engineered sulfamidase
In order to increase sulfamidase secretion from the liver and thus the amount of the enzyme in the blood stream available to specifically target the brain, we engineered the sulfamidase by replacing its own signal peptide (SP) with an alternative one. Two signal peptides have been tested, the Iduronate-2-sulfatase (IDS) signal peptide and the human antitrypsin (AAT) signal peptide. The rationale behind the use of these two signal peptides is that IDS is a lysosomal enzyme that has been demonstrated to be secreted at high levels from the liver while the AAT is a highly secreted enzyme. To test the functionality of chimeric sulfamidase coding sequences containing either the IDS-SP or the AAT-SP we transfected the constructs in Hela and MEF cells derived from MPS-IIIA mice; as control, the cells were also transfected with not-engineered SGSH enzyme. Two days after transfection we measured the SGSH activity in the pellet and in conditioned medium of transfected cells. In the cells transfected with either hAAT(SP)-SGSH-Flag or IDS(SP)-SGSH-Flag, the level of sulfamidase protein detected in the pellet and the medium was increased compared to the level detected in control cells (transfected with the not-engineered sulfamidase) (Fig.1). Such an increase in sulfamidase protein levels was due to both increased efficiency in secretion (medium/total activity; see graph on the right of figure 2) and increased stability of engineered sulfamidase (Fig.1). The analysis of SGSH activity in MEF cells derived from MPSIIIA mice confirmed the results obtained in the Hela cells (not shown).
Engineering the final chimeric sulfamidase. The final goal of our project is to product a modified sulfamidase capable to cross the BBB and target the CNS via receptor-mediated transcytosis. For this reason before starting the experiments aimed at evaluating the therapeutic efficacy of the substituting SP signal in SGSH, we further engineered the modified SGSH with a specific brain-targeting protein domain, the Low Density Lipoprotein receptor (LDLR)-binding domain of the Apolipoprotein B (ApoB LDLR-BD) (Fig.3). The Binding Domain of ApoB will allow the sulfamidase to reach the brain cells by binding LDL receptors, which are abundant on the endothelial cells of BBB. The two final engineered sulfamidase constructs comprise at C-terminal the ApoB LDLR-BD and at N-terminal either an IDS or an hAAT signal peptide (IDSsp-SGSHflag-ApoB and hAATsp-SGSHflag-ApoB) (Fig. 3). To evaluate the functionality of IDSsp-SGSHflag-ApoB and hAATsp-SGSHflag- ApoB we transfected MPSIIIA MEF cells with these final constructs and compared the outcomes with those resulting from the transfections with partial modified (hAATsp-SGSHFlag, IDSsp-SGSH-Flag) and not modified SGSH constructs. Surprisingly, we observed that SGSH activity in the pellet and in the conditioned medium was higher in the cells transfected with the final chimeric constructs compared with the activity measured in the cells transfected with the other constructs, indicating that final engineered sulfamidase were efficiently secreted and even more stable compared to partial engineered sulfamidase (Fig.4). These results were associated with a higher secretion efficiency of the final engineered sulfamidase enzymes with respect to not-engineered sulfamidase. The secretion efficiency of the final chimeric constructs was similar to that measured after transfection of partial chimeric sulfamidase (containing only the alternative signal peptide). In conclusion these results demonstrate that: (i) the chimeric sulfamidase enzymes containing the alternative signal peptide are functional and active; (ii) they are more stable with respect to not-modified sulfamidase; (iii) they are secreted with increased efficiency compared to not engineered sulfamidase enzyme; (iv) the introduction of the ApoB LDLR-BD to produce the final engineered sulfamidase did not affect neither the functionality nor the increased secretion efficiency observed in the cells transfected with the partial engineered sulfamidase. In addition, the final engineered constructs appear to be more stable compared to partial engineered constructs.
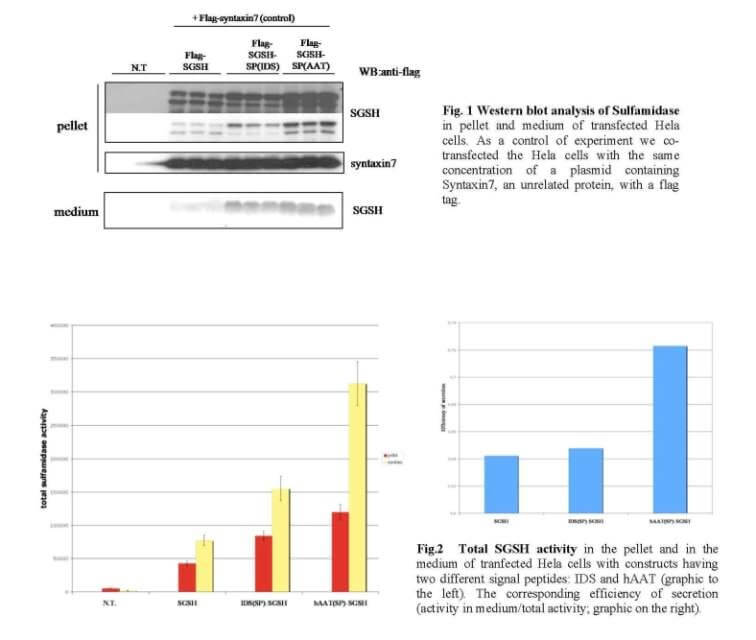
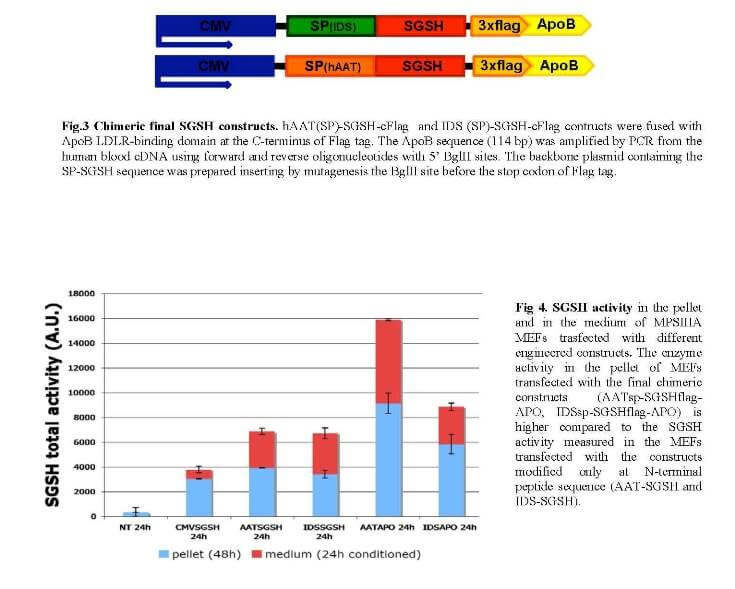
Preliminary in vivo results in MPS IIIA mice injected with final engineered sulfamidase
We obtained very preliminary but extremely encouraging results in MPS-IIIA injected with the final constructs hAATsp-SGSHflag-ApoB. Adult MPS-IIIA mice were systemically injected with AAV2/8-TBG- hAATsp-SGSHflag-ApoB (the TBG is a liver specific promoter). A group of MPS-IIIA were also injected with AAV2/8-TBG-SGSH (containing the not modified sulfamidase) as control. The mice were sacrificed one month after injection. In the mice injected with the chimeric sulfamidase we observed higher liver sulfamidase activity and a very strong increase in the sulfamidase secretion respect to control mice. Moreover, we detected a significant increase in SGSH activity into the brain of mice injected with the chimeric sulfamidase. We are now evaluating the effective cross of BBB by the chimeric sulfamidase. As planned in the proposal, we have also designed an in vivo large study aimed to evaluate the rescue of pathological phenotype in MPS-IIIA. We plan to complete this study within one year.
Dr. Katherine Ponder
Washington University, St. Louis, MO
The role of cathepsin K in cardiac valve disease in MPS
Mucopolysaccharidosis (MPS) is due to a genetic deficiency in the activity of an enzyme that degrades glycosaminoglycans. One of the serious manifestations of MPS is the development of heart disease, which can result in reduced delivery of oxygenated blood to the body. This can involve thickened heart valves that block the flow of blood, and/or heart valves that are leaky and allow blood to flow in the wrong direction. The goal of this project is to understand what causes heart valve problems, which may ultimately allow us to identify a therapy to prevent these heart valve abnormalities from developing.
I. Evaluate collagen and elastin in the extracellular matrix (ECM) of mitral valves, chordae tendineae, and aortic valves in MPS I and MPS VII dogs by light and electron microscopy.
The goal of this aim is to determine if collagen and elastin structure are abnormal in heart valve tissues using histochemical techniques. Collagen is the major extracellular matrix protein of the heart valves. We have demonstrated that the amount of structurally-intact collagen in the mitral valve is approximately 2% of normal at 2 years of age in MPS VII dogs, and that neonatal gene therapy with a retroviral vector can improve the collagen structure. This is consistent with the hypothesis that abnormal collagen structure is responsible for the abnormal valve function. The aortic valves and chordae tendinae have some reduction in structurally intact collagen, but this is less severe. We are in the process of quantifying these results at various ages in different treatment groups.
II. Determine if RNA for cathepsin K or other genes involved in ECM assembly or degradation are upregulated in the mitral valves, chordae tendineae, and aortic valves in MPS VII dogs
Abnormal collagen structure could reflect a failure to synthesize or assemble collagen or elastin properly, or an increase in expression of enzymes with collagenase activity. Mitral valves, chordae tendineae, and aortic valves have been isolated from normal, MPS VII, and retroviral vector-treated MPS VII dogs at 6 months of age, when collagen fragmentation is apparent in untreated MPS VII dogs, and expression of several genes from the mitral valve have been evaluated. Cathepsins and matrix metalloproteinases (MMP) are enzymes that can degrade collagen. RNA analysis demonstrated that cathepsin B, S, and W, and MMP 8, 9, and 12 were markedly upregulated (10- to 100-fold) in the mitral valves of untreated MPS VII dogs. Although cathepsin K RNA was not significantly elevated, several of the other cathepsins and the MMPs have collagen-degrading activity. We are in the process of using microarray analysis to look for changes in expression of other genes that are involved in collagen assembly or degradation, and evaluating the signal transduction pathways that may be involved. This may identify targets that can be inhibited in order to reduce collagen abnormalities.
III. Evaluate enzyme activities and GAG levels in the mitral valves, chordae tendineae, and aortic valves in MPS VII dogs
The final aim of this project will evaluate extracts from the mitral valves, chordae tendineae, and aortic valves of normal, MPS VII, and RV-treated MPS VII dogs for several biochemical parameters. Homogenized samples have been tested and found to have markedly elevated cathepsin activities, although the specific cathepsins that are involved are still being evaluated, and MMP activities have not yet been tested. These data further substantiate our hypothesis that increases in cathepsin activities contribute to abnormal collagen structure, although additional assays still need to be performed.
Dr. Calogera Simonaro
Mount Sinai School of Medicine, New York, NY
Novel anti-inflammatory therapies for the mucopolysaccharidoses
Current therapeutic strategies for the MPS disorders, including enzyme replacement therapy (ERT), have limited effects on the bones and joints. The premise of our research is that new and improved treatment approaches are necessary for these disorders, and that this is best achieved by an improved understanding of the underlying pathogenic mechanisms. Our recent research has focused on the involvement of the toll-like receptor 4 (TLR4) signaling (inflammatory) pathway in MPS bone and joint disease, and the use of anti-inflammatory agents for the treatment of these diseases. The most recent studies were published in the Proceedings from the National Academy of Sciences (Simonaro et al., Proc Natl Acad Sci USA. 2010 Jan 5;107(1):222-7), and will be summarized below.
TLR4 knockout (KO) mice were bred to MPS type VII mice. Inactivation of this proinflammatory pathway in double KO mice corrected many biochemical and clinical features of the MPS disease, suggesting that drugs targeting this pathway could be effective in the treatment of these disorders. Double KO animals grew substantially better than MPS VII mice alone, and had longer and thinner bones. The levels of several inflammatory cytokines, including TNF-?, also were substantially reduced in the double KO mice, leading us to evaluate the effects of the FDA-approved anti-TNF-? drug, RemicadeTM, in MPS VI rats. When initiated pre-symptomatically, intravenous RemicadeTM treatment prevented the elevation of TNF-? and other inflammatory molecules in the blood. Importantly, the levels of these markers also were markedly reduced in cartilage (chondrocytes) and synovial membranes of the treated animals. Although the overall growth of these animals was not improved by RemicadeTM treatment, the number of apoptotic or dead chondrocytes were reduced by ~50%, as was the infiltration of synovial tissue into the underlying bone. These results indicated positive effects of RemicadeTM treatment at the sites of pathology. RemicadeTM treatment also reversed the established inflammatory disease in older animals. Thus, these studies revealed the important role of TLR4 signaling in the pathogenesis of MPS bone and joint disease, and suggested that targeting a downstream mediator of this pathway, TNF-?, might have a positive effect in attenuating the inflammatory response in MPS. This could lead to improved joint/bone pathology and also increase the accessibility of synovial tissues to recombinant proteins, thus improving the efficacy of ERT.
Towards this end, we have recently finished our combination studies in the MPS VI rats with RemicadeTM and ERT (NaglazymeTM), and are in the process of performing a comprehensive evaluation of the effects on the bone and joint disease. We hope that these studies will provide essential data that might fast-track this therapy into the clinic to be evaluated in MPS patients.
Dr. Maria Pia Cosma
TIGEM, Naples, Italy
AV2/5CMV-IDS therapy in MPSII mice: correction of CNS defects through IDS delivery across the blood-brain barrier
Results funded by the MPS society have been published in the following paper:
Correction of CNS defects in the MPSII mouse model via systemic enzyme replacement therapy. Polito VA, Abbondante S, Polishchuk RS, Nusco E, Salvia R and Cosma MP. Human Molecular Genetics 2010, Vol 19, No 24.
Mucopolysaccharidosis type II (MPSII), or Hunter syndrome, is a devastating disorder associated with a shortened life expectancy. Patients affected by MPSII have a variety of symptoms that affect all organs of the body and may include progressive cognitive impairment. MPSII is due to inactivity of the enzyme iduronate-2-sulfatase (IDS), which results in the accumulation of storage material in the lysosomes, such as dermatan and heparan sulfates, with consequent cell degeneration in all tissues including, in the severe phenotype, neuro- degeneration in the central nervous system (CNS). To date, the only treatment available is systemic infusion of IDS, which ameliorates exclusively certain visceral defects. Therefore, it is important to simultaneously treat the visceral and CNS defects of the MPSII patients. Here, we have developed enzyme replacement therapy (ERT) protocols in a mouse model that allow the IDS to reach the brain, with the substantial correction of the CNS phenotype and of the neurobehavioral features. Treatments were beneficial even in adult and old MPSII mice, using relatively low doses of infused IDS over long intervals. This study demonstrates that CNS defects of MPSII mice can be treated by systemic ERT, providing the potential for development of an effective treatment for MPSII patients.
Dr. Jeffrey D. Esko
University of California, San Diego, CA
Substrate Reduction Strategy for MPS IIIA
The original focus of this grant was to demonstrate whether substrate reduction would prove effective as a treatment for Sanfiippo syndromes. As outlined in our progress report 2010, siRNA knock down of the heparan sulfate biosynthetic gene EXT1 in human MPSIIIa fibroblasts is able to diminish lysosomal storage by ~50% ex vivo. To test substrate reduction therapy in vivo, we have crossed MPSIIIa (Sgsh-/-) mice onto an Ext1+/- background. As shown in figure 1a, Ext1 heterozygosity reduced heparan sulfate chain length by ~25% in mouse embryonic fibroblasts (MEFs) isolated from these mice. This reduction in the amount of cell surface heparan sulfate was sufficient to normalize turnover by ~30% (Fig. 1b), demonstrating the efficacy of this substrate reduction therapy approach.
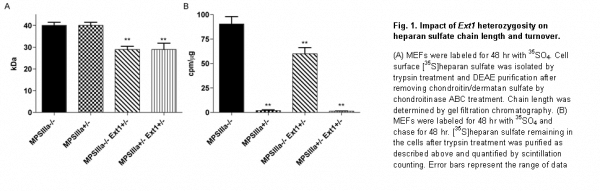
Currently we are quantifying the amount of heparan sulfate in the tissues ofSgsh-/- mice that are heterozygous forExt1 using mass spectrometry to determine the impact of substrate reduction therapy in different tissues. In addition, the histological markers GFAP and Ubiquitin are being used to determine the impact of substrate reduction on astrocytosis and aberrant autophagy in the brain, respectively. To complement these studies we are also breeding MPSIIIa mice heterozygous for bothExt1 andExt2. Double heterozygosity should result in heparan sulfate chains even shorter than those observed inExt1heterozygotes and may have a greater impact on lysosomal storage and pathology. These studies should be completed in the next 3 – 6 months.
In addition to its ability to reduce lysosomal storage, we hypothesized that substrate reduction therapy may serve as a cost effective way to increase the effectiveness of enzyme replacement therapy. To test this, we compared the dose of recombinant sulfamidase necessary to normalize heparan sulfate turnover in MEFs derived fromSgsh-/- orSgsh-/-;Ext1+/- mice. Importantly, the effective dose (ED50) was improved by more than 2- fold in MPSIIIa MEFs heterozygous forExt1, suggesting that combined substrate reduction therapy and enzyme replacement therapy may provide a more efficient approach to treating Sanfilippo disease. To determine the impact ofExt heterozygosity on the sensitivity ofSgsh-/- mice to enzyme replacement therapy in vivo, we are currently quantifying heparan sulfate levels in different organs ofSgsh-/-,Sgsh-/-;Ext1+/ and Sgsh-/-;Ext1+/-;Ext2+/- mice treated with 0, 0.1, 0.3 and 2 mg/kg recombinant sulfamidase using mass spectrometry. These studies should be completed in the 3 6 months.
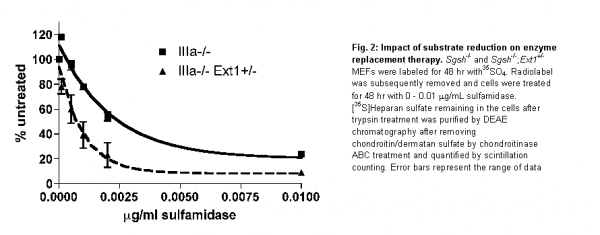
In summary, we are approaching completion of both aims of the grant. Substrate reduction therapy has been demonstrated ex vivo using siRNA and gene knock out of Ext1 and in vivo studies are underway. In addition to the studies outlined above, funds provided by this grant has allowed us to successfully characterize secondary accumulation of dermatan sulfate in Sanfilippo fibroblasts (Lamanna et al.,J. Biol. Chem. 2011). To determine the impact of secondary dermatan sulfate storage on disease pathology, we are characterizing dermatan sulfate levels in different organs ofSgsh-/- mice as well as the sensitivity of secondary storage to enzyme replacement therapy.
We would like to thank the National MPS Society for this funding opportunity.
Dr. Alessandro Fraldi
TIGEM, Naples, Italy
Developing a systemic AAV-mediated gene therapy approach to cross the blood-brain barrier and treat CNS
pathology in Mucopolysaccharidosis type IIIA
Cellular trafficking of the chimeric sulfamidase enzymes
Understanding the cellular trafficking of the chimeric sulfamidase enzymes (containing the alternative signal peptide and the ApoB LDLR-BD) is critical to evaluate the clinical efficacy of the engineered sulfamidase and to correctly interpret the results we will obtain from thein vivo studies. We analyzed the capability of the chimeric sulfamidase enzymes to correctly localize with lysosomal compartment in transfected cells and in cells receiving the enzyme upon uptake. The flag tag was replaced with a myc tag, which give more reliable and specific signal in immunofluorescence analysis. The IDSsp-SGSHmyc-ApoB and hAATsp-SGSHmyc-ApoB along with partial modified sulfamidase enzymes (containing only the alternative signal peptides: IDSsp-SGSHmyc and hAATsp-SGSHmyc) and not-modified sulfamidase (SGSHmyc) were transfected in MPS-IIIA MEFs. Immunostaining with anti-myc and anti-LAMP1 antibodies showed a lysosomal localization for both partial and final engineered constructs similar to that observed in cells transfected with not-modified sulfamidase (Fig. 1).
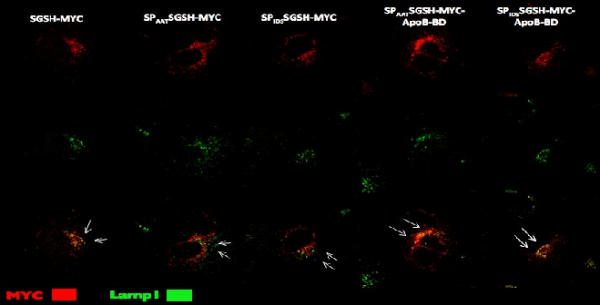
Figure 1. MPS-IIIAMEFcells weretransfectedwith either partial or final engineered constructs or with control not-modifiedSGSHconstruct. All thecontructscontained amyctag. The chimeric constructs display alysosomallocalization as showed byimmunostainingwithanti-mycandanti-LAMP1antibodies.
We then analyzed the capability of chimeric sulfamidase enzyme to be uptaken from MPS-IIIA cells and re-localize to lysosomal compartment. HepG2 cells were transfected with IDSsp-SGSHmyc-ApoB, hAATsp-SGSHmyc-ApoB along with partial modified sulfamidase enzymes (IDSsp-SGSHmyc and hAATsp-SGSHmyc) and non-modified sulfamidase (SGSHmyc). MPS-IIIA MEFs were then incubated with the conditioned medium derived from each transfection. The sulfamidase activity and the subcellular localization of both chimeric and not-modified sulfamidase enzymes were then evaluated in MPS-IIIA MEFs. As shown in figure 2 all the chimeric sulfamidase enzymes display a specific activity in the recipient MPS-IIIA MEF cells thus demonstrating the capability of the chimeric enzymes to be efficiently uptaken (Figure 2). In addition, the chimeric enzymes were also able to correctly re-localize to the lysosomal compartment upon be uptaken (Figure 3).
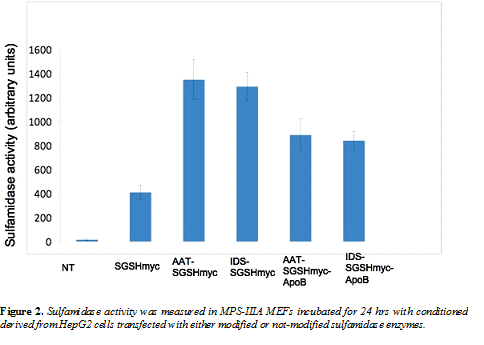
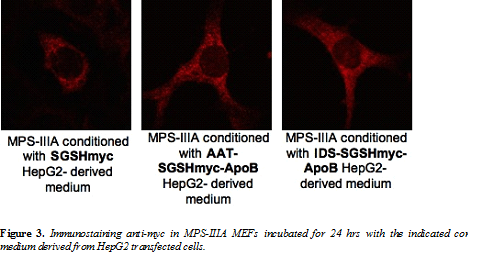
In vivo large study in MPS-IIIA mice
We expanded the MPS-IIIA colony and have obtained a large number of mice to be used in thein vivo large study. We systemically injected one-month old MPS-IIIA mice with AAV2/8-TBG vectors harboring cDNAs encoding either AATsp or IDSsp N-terminal-modified sulfamidase enzymes containing the ApoB-LDLR-BD at their C-terminal (AATsp-SGSH-myc-ApoB-BD and IDSsp-SGSH-myc-ApoB-BD).
Control mice have been injected with AAV2/8 expressing either the not-modified sulfamidase enzyme (SGSHmyc) or the partially modified sulfamidase enzymes (only containing the signal peptide replacement; AATsp-SGSH-myc and IDSsp-SGSH-myc). We established three different time points after injections for the evaluation of CNS phenotype rescue (1 month, 3 months and 7 months). We sacrificed the mice corresponding to the first two time points (1 month and 3 months post-injection). These mice are under evaluation for CNS transduction (enzyme activity into the brain) and CNS pathology (storage, autophagy and inflammation). We obtained very important results by measuring the sulfamidase enzyme activities into the brain of MPS-IIIA mice 3 months post-injection. A stronger sulfamidase activity was observed in the brain of MPS-IIIA mice injected with AAV2/8 encoding the modified sulfamidase enzyme AATsp-SGSH-myc-ApoB-BD when compared to the sulfamidase activity observed in the brain of MPS-IIIA mice injected with not-modified sulfamidase (SGSH-myc) (Figure 4). Moreover, the sulfamidase activity in the brain of MPS-IIIA injected with the modified sulfamidase was also associated to the presence of the enzyme into the brain of injected mice as shown by immunostaining anti-myc (Figure 5).
We are now completing the analysis of the first two groups of injected mice corresponding to 1 month and 3 months post-injection (CNS transduction and CNS pathology).The mice a 7 months post-injection will be assessed for behavioural phenotype at the end of September 2011.
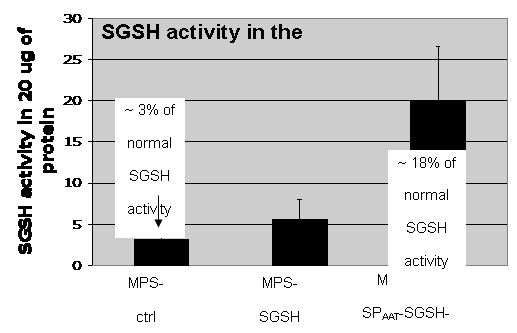
Figure 4.SGSHactivity was measured in the brain ofMPS-IIIAmice systemically injected withAAV2/8-TBG-hAATsp-SGSHmyc-ApoBorAAV2/8-TBG-SGSHmyc. The activity in control not- injectedMPS-IIIAbrain was also displayed.
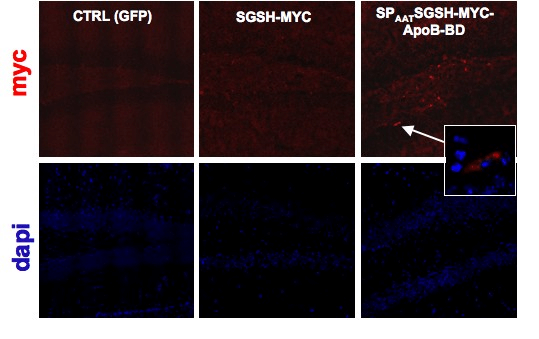
Figure 5.Immunostaining anti-myc in the hippocampus of MPS-IIIA mice systemically injected with either AAV2/8-TBG-hAATsp-SGSHmyc-ApoB or AAV2/8-TBG-SGSHmyc. Control MPS-IIIA hippocampus was also displayed.
Dr. Calogera M. Simonaro
Mount Sinai School of Medicine, New York, NY
Novel Anti-Inflammatory Therapies For The Mucopolysaccharidoses
Although enzyme replacement therapy (ERT) is currently available for three MPS diseases and under development for others, the effects of this therapy on bone and cartilage are very limited. Thus, new approaches are clearly needed to more effectively treat MPS patients, alone or as adjuncts to ERT. In this research project we comprehensively evaluated the bones and joints of MPS VI rats treated by anti-inflammatory therapy (i.e., anti-TNF-alpha therapy), alone and in combination with ERT.
Several anti TNF-alpha drugs (e.g., Remicade, Embrel) are currently used clinically for the treatment of arthritis, inflammatory bowel disease and others common diseases, and the underlying premise of our work is that if these drugs proved effective in MPS animal models, they could potentially be fast-tracked into clinical use for MPS patients. Our previous work also has shown that the anti TNF-alpha inflammatory pathway is activated in many MPS animal models and patients (i.e., the toll-like receptor 4, TLR4, pathway), providing the scientific rationale for this approach.
In our first set of experiments we completed a proof-of-principle experiment in which the TNF-alpha inflammatory pathway (i.e., Toll-like receptor 4 pathway) was inactivated in mice with MPS VII. We found that when this pathway was inactivated from the earliest stages of development, there was a significant improvement in the bone length, bone growth plates, and joint pathology of the MPS VII animals. The paper reporting these findings was published in the Proceedings of the National Academy of Sciences in 2010.
We next turned to experiments evaluating anti TNF-alpha therapy in MPS VI rats, alone and in combination with ERT. In adult MPS VI rats that were treated by anti-TNF-alpha therapy, we found that the circulating levels of many inflammatory molecules in the blood were substantially reduced. Surprisingly, ERT alone also substantially reduced the circulating levels of these inflammatory markers, supporting the concept that the inflammatory pathways in MPS are directly activated by glycosaminoglycan (GAG) storage. We hypothesize that the reduction of circulating inflammatory makers by ERT reflects the delivery and function of the enzyme in readily assessable organs in the MPS animals, such as the liver, spleen, etc. Animals treated by ERT or anti TNF-alpha therapy alone did not exhibit any significant improvement in bone growth. However, the articular (joint) cartilage of animals receiving anti-TNF-alpha therapy had fewer dying (apoptotic) cells, in contrast to animals receiving ERT that were similar to untreated MPS animals. Importantly, when the two treatments were combined, notable clinical and other improvements were observed.
MPS VI rats receiving combined treated exhibited a more normal gait and could remain longer on a rotorod apparatus better than animals receiving either anti TNF-alpha or ERT. Their bones were also slightly longer, and there was much less evidence of inflammation in the joints (e.g., synovial tissue hyperplasia). Perhaps most impressively (and unexpectedly), animals receiving combined treatment had markedly less deformed tracheas, with thinner tracheal walls and wider open spaces. The results of these studies have been summarized in a manuscript that is currently in review. In conclusion, these animal models studies have suggested that combining anti TNF-alpha therapy with ERT may provide substantial clinical benefits to MPS patients. In addition to a direct effect on inflammation, these therapies could also reduce immune responses against the recombinant enzymes, and improve the accessibility of the enzymes to pathologic sites in vivo. Future studies are planned in the dog and cat models of MPS, and using different anti-inflammatory therapies.
2008 Research Grant PDF Download
Over $3 million awarded for research grants since 2000!
The National MPS Society has awarded $499,000 for research grants in 2008. The funding that the Society provides has been and continues to be crucial as we move forward with our mission to find the cures.
Drs. Bigger, Montano, Sands, Serafini, and Steet were awarded the general research grants of $60,000 each. Drs. Brunetti-Pierri and Crawford were each awarded $65,000 for MPS II research, and Dr. Ballabio was awarded the MPS III grant for $60,000. Each grant is for two years, and the researchers will receive half of the total each year.
We received 22 letters of intent from researchers throughout the world for the eight research grants offered in 2008. After reviewing those letters, our Scientific Advisory Board review committee requested full grants proposals from 12 researchers.
This year we collaborated with two foundations to offer an MPS III partnership grant. We did not have enough funds in the MPS III research category to fund a grant, and we are very grateful to the Childrens Medical Research Foundation and Bens Dream Foundation for helping to fund this grant. Opportunities such as this ensure that our research dollars are not dormant for a year as we await additional donations to fund a grant. Its also a wonderful opportunity for the MPS community to join together as we strive to meet our common goal to find the cures.
Money from our research funds supported the expert Newborn Screening meeting held February 1, 2008 and reported in summer 2008 Courage. For the last three years the UK MPS Society has funded the research of Prof. Grzegorz Wegrzyn at the University of Gdansk in Poland, Development of gene expression-targeted isoflavone therapy for MPS III.” The UK requested support this year from our sister organizations in the International MPS Network to fund the extension 4th year. The National MPS Society has funded the requested $4,000 for Prof. Wegrzyns research.
Dr. Nicola Brunetti-Pierri
Baylor College of Medicine, Houston, TX
HDAd gene therapy for lysosomal storage disorders”
Lysosomal storage disorders (LSD) often present with severe neurologic involvement. However, currently available treatments are not effective to treat this significant problem. Both enzyme replacement therapy and gene therapy have failed to show a significant neurologic improvement because the deficient enzyme is transported from the bloodstream to the brain with very low efficiency. To overcome this obstacle, we propose to inject a gene therapy vector directly into the brain fluid (called cerebrospinal fluid or CSF) through a simple and minimally invasive lumbar puncture. By this method, our gene therapy vector will transfer the gene encoding for the deficient lysosomal enzyme to the brain cells lining the CSF spaces. We hypothesize that these cells will secrete the enzyme in the CSF and through the CSF circulation it will diffusely penetrate into the brain to correct the storage disease. The goals of this proposal are to test the efficacy of this approach in mice affected with MPSII and the safety in nonhuman primates (baboons) because large animal models can better predict the outcomes in humans. Therefore, the studies included in this proposal have the potential to generate clinically relevant results which could be applicable for all LSD with neurologic involvement.
Dr. Brett E. Crawford
Zacharon Pharmaceuticals Inc., La Jolla CA,
Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II”
Mucopolysaccharidosis (MPS) is a collection of genetic disorders caused by mutations in genes encoding enzymes required to degrade carbohydrate structures known as glycosaminoglycans (GAGs). The impaired degradation caused by these mutations leads to accumulation of GAGs within cells which in turn leads to serious multi-system disease. Iduronate sulfatase is a critical component of the GAG degradation system, this enzyme is responsible for removing the 2-O-sulfate residues in GAGs that are being degraded. In MPS II patients, the impaired function of the iduronate sulfatase leads to the accumulation of GAG fragments with 2-O-sulfate groups which cannot be degraded. We have discovered compounds that inhibit GAG 2-O-sulfation. These compounds are potentially the starting point for a novel substrate reduction therapy for MPS II. Because these compounds can reduce the amount of 2-O-sulfated GAGs made by cells, it is possible that they could reduce GAG accumulation due to an impaired iduronate sulfatase. In this application, we propose to test these compounds in MPS II models to determine if inhibiting GAG synthesis can reduce GAG accumulation and alleviate symptoms of the disease.
Dr. Andrea Ballabio
TIGEM (Telethon Institute of Genetics & Medicine), Naples, Italy
Modulation of autophagy as a potential therapeutic approach for MPS”
Autophagy is a lysosome-mediated degradation pathway in which large portion of citosol are sequestered in specific vesicles (autophagosomes) and then degraded upon fusion with lysosomes. We demonstrated in two different mouse models of mucoplysaccharisodis, the Multiple Sulfatase Deficiency (MSD) and mucopolysaccharidosis type-IIIA (MPS-IIIA), an impairment of autophagy caused by inefficient fusion between autophagosomes and lysosomes. This results in an abnormal accumulation of different toxic substrates that ultimately lead to cell damage and death. Our results are supported by independent studies demonstrating that a dysfunction of autophagy also occurs in other forms of lysosomal storage diseases. The goal of this project is to exploit novel therapeutic strategies to treat MPS pathology based on the prevention/removal of the toxic substrates that accumulate as a consequence of inefficient autophagic degradation. This will be achieved using both pharmacological and genetic approaches. Our results will be instrumental to develop new therapeutic strategies in human patients.
Dr. Brian Bigger
Royal Manchester Children’s Hospital, Manchester, UK
The effect of heparan sulphate on stem cell homing and engraftment in MPS I”
MPS I Hurler is a fatal genetic disease caused by the lack of a specific enzyme which helps to break down large sugars called glycosaminoglycans (GAGs) in the body. The only treatment is stem cell transplantation, where cells from healthy donors replace patients own bone marrow and produce the missing enzyme. Only around half (56%) of these transplants are successful first time; often a second or third transplant is needed and this can be more risky. In Hurler patients, two types of GAG, heparan sulphate and dermatan sulphate, cannot be broken down, and instead they accumulate inside as well as on the outside of cells. Heparan sulphate helps cells in the body to signal to each other, and is needed by stem cells to home to the bone marrow following transplantation. It is also one of the components of extracellular matrix (ECM); the material that fills the spaces between cells. We have shown that normal cells from a stem cell transplant home differently across ECM from MPS I mice and want to identify if this or other factors cause stem cell transplants to fail. This will help us develop safer transplants for patients with MPS I Hurler.
Dr. Adriana M Montano
Saint Louis University School of Medicine, St Louis, MO
Identification of genes for keratin sulfate biosynthesis: toward development of RNAi mediated therapy”
The main goal of this research is to establish a novel therapeutic system for MPS IVA (Morquio A) by reducing the synthesis of the accumulated substrate (keratan sulfate) in the skeletal tissue to improve the bone lesions. In this proposal, we will test a new approach by partially blocking the synthesis of skeletal keratan sulfate mainly produced in cartilage cells of Morquio A patients. First, two candidate genes responsible for the synthesis of skeletal keratin sulfate will be characterized functionally. Afterwards the enzyme(s) responsible for the synthesis of skeletal keratin sulfate will be attenuated by a recently developed RNA interference method. The targeted gene is suppressed at the RNA level. Our modified RNA interference system will be unique and novel since the therapeutic agent is targeted to the major bone matrix, hydroxyapatite, by attaching a short acidic peptide to the agent. The attenuation of synthesis of the enzyme(s) will be tested initially in vitro in cartilage cells of MPS IVA patients. Successive preclinical trial on animal model(s) will provide critical information leading to human clinical trials. This new approach could be applicable to all types of MPS which store different types of glycosaminoglycans and suffer from bone lesions.
Dr. Mark S. Sands
Washington University School of Medicine, St. Louis, MO
Metabolic adaptations and phenotypic consequences of blocking lysosomal recycling”
Lysosomal storage diseases (LSDs) are caused by a deficiency of enzymes responsible for recycling material in cells. This recycling serves an important purpose by saving the cell energy. In LSDs recycling is interrupted and storage material builds up in lysosomes, likely contributing to disease. The amount of energy saved by a normal cell by lysosomal recycling is equivalent to the amount of inaccessible energy (storage material) in the lysosome of an affected cell. This can be an enormous amount of energy over the life of a cell. Since lysosomal recycling is blocked in LSDs, an affected cell has to expend more energy to carry out its normal functions in order to make up for the inaccessible energy stored in the lysosome. In an organism, this will result in a deficiency in fat stores. We previously documented this effect in five different mouse models of lysosomal storage disease. We are currently conducting experiments in mice to ascertain what adaptations the affected cells are making to the lack of recycling and how this contributes to the symptoms and progression of the disease. The knowledge gained from these studies will be used to design and test the effectiveness of dietary interventions.
Dr. Marta Serafini and Dr. Ettore Biagi
Dulbecco Telethon Institute at M.Tettamanti Research Center Clinica Pediatrica Univ., Monza, Italy
Marrow mesenchymal stem cell therapy for MPS I”
Affecting one in 100,000 children, Hurler syndrome is a rare genetic disorder where the IDUA enzyme, which normally breaks down the mucopolysaccharides dermatan and heparan sulphate, is missing. These mucopolysaccharides build up in all tissues in the body causing progressive deterioration and abnormal function of multiple organs. Hematopoietic cell transplantation (HCT) is one of the most promising treatments available that retards the progression of the disease. The clinical success of HCT as therapeutic approach for MPS I is compromised by the high frequency of graft rejection, incomplete donor chimerism and by inefficiency to prevent and correct skeletal abnormalities associated with the disease. This proposal aims at investigating if the use of supplemental stem cell therapy can improve the efficiency of HCT. Our research interest is focussed on a population of stem cells called mesenchymal stem cells (MSC), which can significantly contribute to regenerate tissues of the mesenchymal lineages, as stroma, bones and cartilage. We want to determine if MSC isolated from healthy donors may facilitate hematopoietic repopulation and skeletal tissues repair in a NOD/SCID/MPS-I mouse model. This initial experience will serve to ascertain if our hypothesis is robust and meet all the criteria to transfer this therapeutic strategy into clinical intervention.
Dr. Richard Steet
University of Georgia Research Foundation, Athens, GA
Investigation of the cartilage pathogenesis of ML II and MPS”
While many tissues throughout the body are affected in individuals with MPS and MPS-related disorders, the central pathology in these patients is observed in bone and joints. Impaired development and progressive destruction of cartilage leads to many debilitating symptoms for MPS patients. The series of molecular events that lead from the primary genetic defect of MPS disorders to the characteristic bone and cartilage pathologies remains poorly understood. Defining these molecular events is important since it will point to new ways to treat these diseases without having to replace the defective enzymes or genes. Our laboratory has been using the zebrafish model system to study the cartilage defects associated with the MPS-related disorder, mucolipidosis II (ML-II). Our current evidence suggests that these cartilage defects are accompanied by changes in the expression level of several types of proteases, enzymes that can degrade extracellular proteins and cause damage to cartilage. We are planning to use zebrafish models of selected MPS and MPS-related disorders to directly test the role of these proteases.
These studies will provide new insight into the disease process of MPS disorders and will serve to identify new targets for therapy.
Dr. Sara Cathey
Greenwood Genetics Center, North Charleston, SC
Natural history study for mucolipidosis
One year partnership grant with ISMRD.
Received 7-09
The Mucolipidosis Project of the Greenwood Genetic Center (GGC) began in 2005. The goals of the project are to establish databases of clinical and laboratory information about ML II and ML III alpha/beta, understand the disease course, provide natural history information to families and the medical community, and raise awareness of the mucolipidoses. These goals are being met and exceeded! The largest group of individuals with ML II and ML III alpha/beta ever reported has been described in the medical literature. Families and doctors have a resource for reliable information. Scientists have access to samples for research. The ML Project is ongoing, allowing the natural course of the diseases to be more fully understood. Fifteen families attended the first ML Clinic at GGC in 2006. The majority of those families, plus others, will participate in GGCs Second ML Clinic, July 27-29, 2009. This is an opportunity to evaluate individuals over time. We can not only study disease progression, but also assess the impact of interventions. Understanding the natural course of disease builds the framework for successful treatments. What interventions work best, and when? Are there markers? of disease in the blood or urine that can be followed to guide therapies? Who are the best candidates for surgery? Which surgeries? By supporting work like the ML Project, the National MPS Society continues to foster research, progress, and hope. Families remain the driving force of this incredible momentum. Your support and participation make great things possible.
Nicola Brunetti-Pierri, M.D.
Department of Molecular and Human Genetics
Baylor College of Medicine, Houston, TX
HDAd gene therapy for lysosomal storage disorders?
Received 7-09
Correction of the neurological manifestations of lysosomal storage diseases has been elusive so far. The goal of our project is to develop a safe and effective strategy for correction of the central nervous system manifestations of Mucopolysaccharidosis II which could be potentially applicable to other lysosomal storage diseases as well.
Helper-dependent adenoviral (HDAd) vectors are devoid of all viral genes and result in long term transgene expression in the absence of chronic toxicity. Because of their ability to infect nondividing cells, including cells of the central nervous system, HDAd vectors are particularly attractive for brain-directed gene therapy. Using a vector expressing a reporter gene (the green fluorescent protein), we have showed that HDAd vectors administered via intratechal injection into the cerebrospinal fluid (CSF) resulted in minimal systemic toxicity and extensive transduction of neuroependymal cells as well as neuronal cells. Importantly, the expression has lasted for at least 3 months and we are planning to look also at longer time points (6 months- 1 year). Given the encouraging results obtained with the reporter gene, we are perfroming experiments in a mouse model of Mucopolysaccharidosis II injecting a vector encoding for the therapeutic gene. These experiments are crucial because they will clarify whether our approach will allow the correction of the central nervous system manifestation in the mice affected with Mucopolysaccharidosis II.a
Brett E. Crawford, Ph.D.
Zacharon Pharmaceuticals Inc., La Jolla CA, 92037
“Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II”
Received 7-09
Thanks to the support of the National MPS Society, we have made important progress during the first year of our research project. As proposed, our work has focused on two aspects of developing a substrate reduction therapy for MPS II: i) the development of methods to quantify GAG accumulation in cultured human MPS cells and ii) the testing of candidate GAG inhibitors in this model system.
In order to develop methods to quantify GAG accumulation in cultured MPS cells, we obtained cells from patients with MPS II from the National Institute of General Medical Sciences Human Genetic Cell Repository. With these cells we developed improved methods to quantify GAG accumulation in cultured MPS cells (the Sensi?Pro Substrate Assay). The Sensi?Pro assay was validated by successfully detecting reductions in GAG accumulation in MPS II cells treated with recombinant iduronate sulfatase. By developing and validating these methods, we now have an experimental model to test the effectiveness of candidate drugs by measuring their ability to impact GAG accumulation in MPS cells.
We have recently found that the assay could also be used clinically as it has the sensitivity to detect GAG accumulation in urine, serum, and cerebral spinal fluid of patients with MPS with several advantages over the alternative methods. Using the Sensi?Pro assay, we have screened our candidate GAG biosynthesis inhibitors to determine if any of these compounds reduce GAG accumulation in MPS II. Through these studies we have identified a series of compounds that reduce GAG accumulation in MPS II cells.
These exciting and positive results provide a foundation to accomplish the next stage of the research project: testing the most promising candidate drugs in the mouse model of MPS II. We have obtained the MPS II mouse model (strain courtesy of Joseph Muenzer, MD, PhD) and are well positioned to accomplish the next stage of the research project.
Dr. Andrea Ballabio
TIGEM (Telethon Institute of Genetics & Medicine), Naples, Italy
Modulation of autophagy as a potential therapeutic approach for MPS?”
Received 7-09
We recently generated new insights in the pathogenic mechanisms of mucoplysaccharidoses and more in general of lysosomal storage disorders. We demonstrated that a crucial lysosomal degradative pathway, autophagy, is severely impaired in two mouse models of mucopolysaccharidoses, Multiple Sulfatase Deficiency (MSD) and mucopolysaccharidosis type-IIIA (MPS-IIIA). As a consequence of a defect of autophagy we observed an abnormal accumulation of several autophagic substrates such as polyubiquitinated aggregates, p62 protein and dysfunctional mithocondria, that lead to cell death and tissue pathology in both models analyzed. During the first year supported by MPS funding, we began to evaluate the therapeutic effect of preventing/removing autophagic-dependent accumulation of toxic substrates in MSD and MPS-IIIA mouse models. We used both a drug delivery approach based on rapamycin administration and a genetic approach based on crossing affected mice with p62 KO mice.
Newborn (0-2 gg) and adult (3 weeks of age) MSD and MPS-IIIA mice received intraperitoneal injection of rapamycin (20mg/kg) three times a week during all treatment period. Mice were analyzed at two time points: 1) an early time point (5 weeks of age) for biochemical and histological analysis (both MPS-IIIA and MSD mice) and 2) a late time point for biochemical, histological and behavioral analysis. We chose 20 weeks of age as late time point for MPS-IIIA mice and 12 weeks of age as a for MSD mice because the most significant difference between normal and affected mice in terms of behavior were observed at this age.
The first group of treated mice (either newborn- or adult-treated mice) were analyzed at 5 weeks of age (early time point) and collected tissues (brain and liver) assessed for the accumulation of p62, alfa-synuclein and polyubiquitined proteins. In all affected mice (MSD and MPS-IIIA) the injection with rapamycin resulted in a significant decrease of toxic accumulation when compared to control PBS-injected affected mice. Importantly, the extent in the decrease of toxic accumulation was higher in mice treated at birth compared to mice treated at 3 weeks of age. We are now evaluating mitochondrial dysfunction and apoptotic cell death in the same tissues. These first data are very encouraging. The other experimental groups of mice (MSD and MPS-IIIA mice injected with rapamycin along with control normal and affected mice PBS-injected) will be analyzed at 20 weeks (MPS-IIIA) or at 12 weeks of age (MSD) to evaluate if the rapamycin treatment is able to recover a normal behavior in affected mice.
We plan to complete this task within 1 year.
The conditional p62 KO mice have been received from Dr Tanakas group (Laboratory of Frontier Science, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan; Komatsu et al., 2007). These mice are now available in our animal facility and the colony has been established and expanded. These mice will be crossed with either MSD or MPS-IIIA mice. We plan to induce the ablation of p62 in specific tissues (brain and liver) to evaluate to which extent the abnormal accumulation of this substrate is involved in the tissue pathology and consequently if its ablation could prevent the pathology itself.
We plan to complete this task within 1 year.
Brian Bigger PhD
MPS Stem Cell Research Group
University of Manchester, Royal Manchester Children’s Hospital
The effect of heparan sulphate on stem cell homing and engraftment in MPS”
Received 7-09
Heparan Sulfate: A Helping Hand or A Sticking Point? Heparan sulfate is stored in the cells of MPS I, II, III, and VII patients. In unaffected cells, heparan sulfate has a very important role in cell to cell signalling: allowing cells to talk to each other by interacting with molecules on the surface of nearby cells, or released into the space between cells. Like all the glycosaminoglycans or GAGs stored in the MPS diseases, it consists of a protein core with large chains of sugars attached to it. The sugar chains can have sulfate groups attached in a variety of patterns, which give them patches of negative charge. The patterns of sulfation differ between different kinds of cells and organs across the body. It is believed that these changing patterns of sulfation are important in deciding which signals get processed by the cell, and which signals get ignored. This work focuses on MPS I Hurler, and bone marrow transplantation. When bone marrow is transplanted, the cells enter the blood stream, and need to find their way to the bone marrow by following signals coming from the bone marrow, like a homing beacon. It is known that heparan sulfate is involved in mediating the homing signal, and we are investigating whether this interaction is altered in MPS I Hurler, and whether this might be influencing the success of bone marrow transplantation for MPS I Hurler, and potentially other MPS diseases. It is well known that large amounts of heparan sulphate and other glycosaminoglycans are stored inside the cells of MPS patients, but we wanted to prove that heparan sulphate is found on the outside of MPS I Hurler cells, where it could take part in signalling. The pictures below show an excess of heparan sulfate, (shown in green) on the surface of bone marrow stromal cells taken from mice with MPS I Hurler. Having shown that there is more heparan sulfate on the cell surface, specialised antibodies were used to show that this heparan sulfate had different sulfation patterns to heparan sulphate from unaffected cells. We found that MPS I Hurler heparan sulfate carries more of the type of sulfation that tends to lead to increased signalling. Indeed, we found that bone marrow cells could home across matrix (the material that fills the gaps between cells) containing MPS I Hurler heparan sulfate almost twice as well as across matrix from unaffected cells. In summary, so far we have found MPS I Hurler bone marrow stromal cells accumulate heparan sulfate externally as well as internally, Heparan sulfate from MPS I Hurler cells has a specific structure that favors homing to bone marrow and alters cell:cell signaling. As this work continues, we hope to clarify the precise role of this altered heparan sulfate in the bone marrow homing and engraftment process, and find ways of manipulating this pathway to improve transplant success.
Adriana M Montano, PhD
Saint Louis University
School of Medicine – Dept of Pediatrics, St Louis, MO
Identification of genes for keratin sulfate biosynthesis: toward development of RNAi mediated therapy?”
Received 7-09
Mucopolysaccharidosis IVA (Morquio A disease) is caused by the deficiency of the enzyme N-acetylgalactosamine-6-sulfate sulfatase (GALNS). The lack of this enzyme leads to the accumulation of undegraded glycosaminoglycans (GAGs): keratan sulfate (KS) and chondroitin-6- sulfate (CS) in all the cells of the body, especially in skeletal tissue.
Several treatment approaches have been focused on providing the deficient enzyme to Morquio A mouse models. Our approach is to partially block the synthesis of KS mainly in cartilage cells to stop the progression of the disease by attenuating one or more genes involved in the synthesis of KS (See the figure).
The complete set of genes involved in the synthesis of KS is unknown posing an obstacle for understanding the biochemical processes leading their accumulation. In order to overcome this problem, we have performed a detailed differential expression analysis and identified two candidate genes that may be involved in the synthesis of KS, and made progress towards their characterization. We have cloned the genes in an expression vector to study their effects in-vitro. In addition, we have cloned the genes in another vector that will facilitate the purification of the proteins resultant as the product of these genes. In-vitro analysis and the protein purification experiments are in progress. After the completion of these crucial experiments, we will focus, in the second year of our program, on (i) the suppression of these genes at the RNA level, and (ii) performing in-vitro tests in cartilage cells of Morquio A patients. The success of the second phase will provide a promising therapy technique for MPS IVA patients.
Mark S. Sands, Ph.D.
Washington University School of Medicine
Department of Internal Medicine, St. Louis, MO
Metabolic adaptations and phenotypic consequences of blocking lysosomal recycling?”
Received 7-09
Specific Aims:
1) Determine the effects of reduced lysosomal recycling on the energy imbalance in affected cells.
2) Determine the effects of dietary intervention on the progression of disease.
Summary:
We generated preliminary data showing that there was a significant energy imbalance of unknown etiology in 5 murine models of different lysosomal storage disease (Woloszynek et al., 2007, J. Biol. Chem., 282:35765). The energy imbalance manifests as a significant decrease in adiposity. These data suggest that lysosomal storage disorders are diseases of deficiency as well as excess (lysosomal storage). We hypothesized that the energy imbalance was due to an increase in energy-intensive reactions required to maintain homeostasis in the context of a decreased pool of metabolites from lysosomal recycling. The goal of this research was to provide additional data supporting this hypothesis and determine the effects of an energy-rich diet on disease progression. We performed an unbiased metabolomics survey of the liver of MPS I animals. This analysis showed that simple sugars, nucleotides and lipids were decreased in the MPS I liver compared to normal. This supports the hypothesis that the animals are in a state of deficiency. The metabolomics analysis also showed an increase in amino acids, amino acid derivatives, dipeptides, and urea. These data suggest that increased protein catabolism is at least partially fulfilling intermediary metabolism. When MPS I animals were placed on a high fat, simple sugar diet (high fat diet) for 4 weeks, most of the abnormal metabolite levels approached normal. Consistent with the apparent increase in protein catabolism, we observed an increase in a marker of autophagy (LC3) in the livers of both MPSI and MPS VII animals. Autophagy was decreased in the livers of MPS I animals placed on the high fat diet, however, the decrease was not statistically significant (p=0.08). Interestingly, autophagy was significantly reduced and approached nearly normal levels in the livers of MPS VII animals maintained on a high fat, simple sugar diet from weaning. MPS VII animals maintained on the high fat diet from weaning had significantly increased body weight for the duration of the study (7 months). Unfortunately, there was no significant improvement in longevity, retinal function or cardiac morphology or function. These data strongly support the hypothesis that animals with LSDs are suffering from nutritional stress similar to starvation. Although a crude high fat, simple sugar diet resulted in only minimal clinical improvement, a more targeted nutrient approach may yet prove beneficial as an adjunct therapy as the mechanism of this energy imbalance is better understood. The data generated from this MPS Society-funded project is currently under review at the Journal of Biological Chemistry.
Year 2 funding:
We have made significant progress on this important area of research during the first year of funding. We are currently breeding the MPS I and MPS VII mutations onto the ATG5- and ATG6 (Beclin)-deficient animals. These gene knockouts interrupt normal autophagy. These double mutant animals will allow us to determine if the changes in autophagy in response to the energy imbalance are helpful or harmful.
Marta Serafini, PhD
STEMMPS Unit, Dulbecco Telethon Institute at M.Tettamanti Research Center Clinica Pediatrica Univ. Milano-Bicocca, Monza, Italy
Marrow mesenchymal stem cell therapy for MPS I?”
Received 7-09
The goal of our project is the identification of a novel cell therapy strategy capable of effectively alleviating disease manifestations, in particular at the skeletal level, still affecting MPS-I children after allogeneic hematopoietic stem cell transplantation.
To reach this goal, we are focusing on the following different aspects:
1. Optimize the culture of human mesenchymal stem cells (hMSC), a population of multipotent stem cells with interesting therapeutic potential and compare hMSC derived from healthy and MPS-I children of similar age.
We defined a new protocol to isolate hMSC clones and compared the properties of our cultures kept under hypoxic/normoxic conditions. We focused on the establishment and characterization of MSC lines from healthy young donors (age 1-10) and MPS-I patients (age 1-3). Lines have been maintained in long-term in vitro culture. We are particularly interested in the capacity of these cells to differentiate into the osteogenic lineage, as our approach aims to demonstrate the repair of MPS-I problems at the skeletal level. For this reason, we compared the in vitro osteogenic differentiation of the generated populations in a time course, monitoring the formation of mineralized matrix and the osteogenic transcription profile. The comparison of osteogenic properties between cells derived from healthy donors and MPS-I patients did not reveal significant differences. Ongoing experiments will evaluate within a few weeks if both the populations are able to establish the bone microenvironment in vivo.
2. Explore the potential of umbilical cord blood (UCB-MSC) and amniotic fluid (AF-MSC) as alternative sources of hMSC,.
Few reports have described an advantage of fetal hMSC in osteogenic differentiation potential over adult hMSC, which may represent optimal sources for bone repair and regeneration. We are currently working on isolation procedures to improve the success rate, which is 20% for UCM samples and 60% for AF samples. We fully characterized the populations obtained and compared their capacity to differentiate into mature osteoblasts and produce a mineralized matrix under permissive osteogenic conditions. The next step of our work will be the evaluation of our transplant experiments in mice to assess their osteogenic capacity.
3. Optimization of hMSC transduction with lentiviral vectors (LV).
To track hMSC transplanted in the NOD/SCID/MPS-I mice, we inserted the GFP reporter gene into hMSC. For this reason, GFP-transduction of hMSC was optimized, tuning vector dose and number of rounds of transduction. According to our generated protocol, we genetically modified the cells with an HIV-1-based lentiviral vector carrying the eGFP reporter gene (in collaboration with Dr. A.Biffi). Efficient GFP expression (range: 87-95% GFP positive cells) was observed three days after transduction and the percentage of GFP-expressing cells remained virtually unchanged in subsequent 10 passages, which correlated to 35 to 40 days post-transduction. We observed that LV-transduced hMSC have a lower proliferation capacity compared to untransduced cells.
To further evaluate whether lentiviral transduction altered the differentiation properties of hMSCs in vitro, we analysed the cells for the typical markers CD73, CD105, CD90 and CD146. We also induced the transduced cells to differentiate into cells of the mesenchymal lineages: adipocytes, osteocytes and chondrocytes. The data obtained are comparable with those from untransduced hMSC. In addition, chromosomal stability of the transduced cells was confirmed by karyotype analysis.
4. Establishment and optimization of the transplant protocol to evaluate the contribution of hMSC to MPS-I disease model.
We are comparing different transplantation protocols in the mouse model MPS-I in order to identify the one allowing for efficient MSC homing to the skeleton and phenotypic amelioration. In fact, for MSC trafficking experiments, the timing of delivery, number of cells delivered and site of MSC infusion may impact the engraftment efficiency and the destination of exogenously delivered cells. Completion of the work dedicated to the assessment of the feasibility and therapeutic efficacy of MSC transplantation in MPS-I mice is expected in the next few months. Through a direct collaboration with Dr. Gupta, we are scheduled to begin the transplant experiments in September 2009.
Richard Steet, Ph.D.
Assistant Professor of Biochemistry and Molecular Biology Complex Carbohydrate Research Center
University of Georgia Research Foundation, Athens, GA
Investigation of the cartilage pathogenesis of ML II and MPS?”
Received 7-09
The pathogenic mechanisms that underlie the bone and cartilage phenotypes in MPS and MPS-related disorders are only now beginning to emerge. Defining these mechanisms is important since it will potentially aid in the development of new therapies. Our proposal aims to take advantage of the power and speed of the zebrafish system to identify factors that contribute to the cartilage pathogenesis in mucolipidosis-II and directly test their contribution to the disease process. These studies will utilize our newly characterized zebrafish model for ML-II and will serve as a prelude for therapeutic development. We are also attempting to model additional MPS and MPS-related disorders in this model organism in order to better understand how loss of specific hydrolases results in the phenotypes noted in patients. Over the past year, we have analyzed the gene expression profiles in wild type and ML-II whole zebrafish embryos and isolated zebrafish chondrocytes, demonstrating that several genes relevant to bone and cartilage homeostasis appear to be upregulated, including cathepsins and matrix metalloproteinases. These findings have been confirmed in chondrocyte and fibroblast-like synoviocyte samples from the feline ML-II model. Unlike MPS cartilage and synoviocyte cells, however, we have not seen increases in genes that express pro-inflammatory cytokines. These preliminary findings may suggest unique mechanisms of cartilage damage in ML-II that are independent of inflammation. Our goal in the coming year will be to reduce the expression of the upregulated proteases in the ML-II zebrafish and assess whether this reduction can suppress the cartilage phenotypes in our model. We are also actively investigating the mechanisms that lead to upregulation of these potentially damaging proteases. Attempts have been made to generate models for other MPS disorders using the zebrafish system. As an entry point to this work, the activity of the GAG-degrading enzyme, beta-glucuronidase, was depleted in zebrafish embryos using antisense oligonucleotides called morpholinos.
Although we were able to effectively suppress beta-glucuronidase activity to less than 5% of wild type levels, no obvious phenotypes were observed in these mutant embryos. This finding has prompted us to examine the lysosomal biology of zebrafish in greater detail. To do so, we determined the expression (by RT-PCR) and the activity (using fluorescent substrates) of several lysosomal hydrolases across a developmental timeline (0-5 days post-fertilization). Our results indicate that, unlike enzymes involved in the breakdown of protein-bound oligosaccharides, expression of GAG-degrading enzymes – such as alpha-iduronidase and beta-glucuronidase – does not significantly increase until later developmental stages (4-5 dpf). By this stage, many of the vital organ systems have already developed in the embryos.
This fact, along with the possibility that GAG accumulation and turnover is relatively slow in developing zebrafish tissue, may limit our ability to model certain MPS disorders in this organism using our current antisense-based approach. Thus, we are now actively seeking new ways to genetically target these hydrolases in order to generate zebrafish mutants with sustained loss of enzyme activity.
2008 Second year reviews
In 2008 the National MPS Society awarded two MPS II grants, one MPS II grant and five general MPS research grants.
Dr. Nicola Brunetti-Pierri
Telethon Institute of Genetics and Medicine
Naples, Italy
HDAd gene therapy for lysosomal storage disorders
Correction of the neurological manifestations of lysosomal storage diseases has been elusive so far. The goal of our project was to develop a safe and effective strategy for correction of the central nervous system manifestations of MPS II which could be potentially applicable to other lysosomal storage diseases as well.
Helper-dependent adenoviral (HDAd) vectors which are devoid of all viral genes can infect post-mitotic cells, including cells of the central nervous system (CNS) and are particularly attractive for brain-directed gene therapy. We have showed that intrathecal injections of HDAd resulted in extensive transduction of ependymal cells and sustained expression of the transgene up to one year post-administration. We have also demonstrated, for the first time, the ability of HDAd injected by this route of delivery to transducer neuronal cells. The transduced neuroepithelial cells can be potentially used to secrete therapeutic proteins into the cerebrospinal fluid (CSF) and provide them via cross-correction to non-transduced cells. Targeting of neuronal cells and long-term transgene expression make this approach attractive for the treatment of several neurologic diseases, including lysosomal storage disorders. An HDAd vector encoding the iduronate sulfatase has been injected by intrathecal injection in a mouse model of Mucopolysaccharidosis II and we are currently processing brain samples to investigate whether correction of central nervous system manifestations has occurred.
Dr. Brett E. Crawford
Zacharon Pharmaceuticals Inc., La Jolla CA,
Glycosaminoglycan inhibitors as substrate reduction therapies for MPS II
With the support from the National MPS Society, we have made significant progress toward developing a new approach designed to treat both the neurological and non?neurological symptoms of MPS. This approach is based on drugs which modify GAGs so that the deficient enzyme is not required to degrade them. Our progress has moved through three important stages of drug development: i) development of methods to quantify disease in human MPS cell samples, ii) testing of candidate drugs in this model system, iii) testing of candidate drugs in mouse models of MPS. The following is a brief description of our progress:
Development of methods to quantify disease in cultured human MPS cells.
The first goal of our proposed research was to develop a laboratory system based on cells from patients with MPS that would allow us to test potential treatments in the lab. A testing system like this is essential to discovering new drugs which can modify GAGs so that the deficient enzyme is not required to degrade them. We were very successful in this aim with the development of the Sensi?Pro Assay. This assay can sensitively and specifically measure the amount of GAGs that have accumulated in the lysosomes of MPS patient samples or cells. While we originally developed the Sensi?Pro Assay to guide our own drug development efforts, we found (through a series of collaborations with MPS experts) that the assay was also very useful in other ways. We have found that it can also detect MPS disease in urine, tissue, blood, and cerebrospinal fluid in addition to human cells. The assay has also been found to be very useful in measuring treatment response, thus it can help in the development of other new treatments and can help clinicians monitor MPS patients and optimize treatment decisions. We are also conducting experiments to show how the assay could be used as a newborn screen for MPS.
Testing of candidate treatments in the Sensi?Pro Assay:
With this testing system developed, we then tested over 100,000 drug candidates and found several promising drug candidates which reduce the lysosomal accumulation of GAGs in MPS I, II, and III (A, B, and C). This exciting milestone demonstrated the principle of modifying GAGs so that the deficient enzymes are not required to degrade them. In order to move these drug candidates toward clinical testing, we first need to improve their characteristics. To accomplish this, we began synthesizing and testing a large number of derivatives of the drug candidates. Through this ongoing effort, we have improved the characteristics of the drug candidates, and the most effective candidates are currently being evaluated for properties that are required for a safe and effective therapy.
Testing of drug candidates in mouse models of MPS:
We obtained the mouse models of MPS I, II, and IIIA. We originally proposed to test our compound in the MPS II mouse, but our MPS II mouse colony is not large enough yet for testing drug candidates. Because our treatment is also effective in MPS IIIA, we tested the most promising drug candidate in mice with MPS IIIA. While this initial study is small, we were very excited to find that the lysosomal accumulation of GAGs was reduced in the brains of the MPS IIIA mice. Also, the mice displayed no adverse effects from treatment over the 20 day study. Larger efficacy studies in MPS I, II, and III are planned for the near future. We are also happy to report that we have recently been awarded an NIH Small Business Innovative Research grant to further our efforts to develop a new approach to treating MPS. We are thankful for the ongoing support of the MPS Society which is helping fund the development of this new approach to treating MPS.
Dr. Andrea Ballabio
TIGEM (Telethon Institute of Genetics & Medicine)
Naples, Italy
Modulation of autophagy as a potential therapeutic approach for MPS
Recent studies highlight the prominent role of inefficient autophagic-dependent mitochondria dysfunction in the pathogenesis of neurodegenerative autophagic diseases as Huntington. Mitochondria are organelles recognized as central players in cell death. Proper recycling of dysfunctional mitochondria relies on PINK1/Parkin -dependent selective autophagy (a process known as mitophagy) that avoids the release of pro-apoptotic factors. These studies prompted us to investigate the role of mitochondria in the pathogenesis of MPSs. Our recent findings demonstrated that dysfunctional mitochondria accumulate, together with other toxic substrates such as p62-aggregates and poly-ubiquitinated proteins, as a consequence of autophagic stress in two mouse models of mucopolysaccharidoses, MSD and MPS-IIIA.
During the second year of MPS funding we decided to investigate more in depth the role of the lysosomal-mitochondrial axis dysfunction in the cellular pathology of both MSD and MPS-IIIA mice. Moreover, as planned in our proposal, we evaluated the recovery of normal mitochondrial function in the brain and liver of MDS and MPS-IIIA mice treated with rapamycin.
We observed that dysfunctional mitochondria accumulate, in a different fashion, in liver and brain from the MPS mouse models analyzed. These organelles showed reduced membrane integrity, low ATP content and significant morphological changes (being fragmented in brain and giant in liver). Such morphological/functional changes lead to cytochrome c release and apoptotic cell death in the liver of the affect mice. We also analyzed the mitochondrial function in the MSD and MPS-IIIA mice treated with rapamycin. After 5 weeks of treatment we observed a significant reduction of dysfunctional mitochondria together with a partial rescue of the pathological phenotype in treated mice. We have obtained the following results:
1) The mitochondria recovered a normal morphology in affected mice treated with rapamycin.
2) We did not detect any release of cytochrom c from liver tissue of affected mice treated with rapamycin, thus meaning that rapamycin successfully enhances the clearance of damaged mitochondria.
3) We have also detected a reduction in the levels of pro-inflammatory cytokines in treated mice. This could be due to either the removal of toxic substrates (aggregate proteins and dysfunctional mitochondria), or to rapamycin immunosuppressive activity, or both.
Together our data provide new evidence that the mitochondrial-lysosomal axis differentially contributes to neurodegeneration and to the systemic features in severe models of mucopolysaccharidoses and that induction of autophagy could be a feasible therapeutic approach for this type of inherited disorders. Importantly, our data need to be extended to evaluate whether the rapamycin treatment was indeed effective in rescuing neurodegenerative processes and behavior phenotype in the affected mice.
Dr. Brian Bigger
Royal Manchester Children’s Hospital
Manchester, UK
The effect of heparan sulphate on stem cell homing and engraftment in MPS I
Introduction
When stem cells are transplanted into patients, they must find their way from the bloodstream into the bone marrow by following signals called chemokines, which are produced by the bone marrow. In recent years, it has become clear that chemokines interact with glycosaminoglycans, heparan sulphate in particular, in order to function normally. Heparan sulphate (HS) is one of the stored molecules in MPS I, as well as MPS II, MPS III and VII. Here, we examine how HS accumulation in MPS I could be affecting homing of stem cells to the bone marrow during bone marrow transplantation, and thus contribute to graft failure.
Year 1: MPS I Bone Marrow Cells Accumulate Excess Highly Sulphated Heparan Sulphate
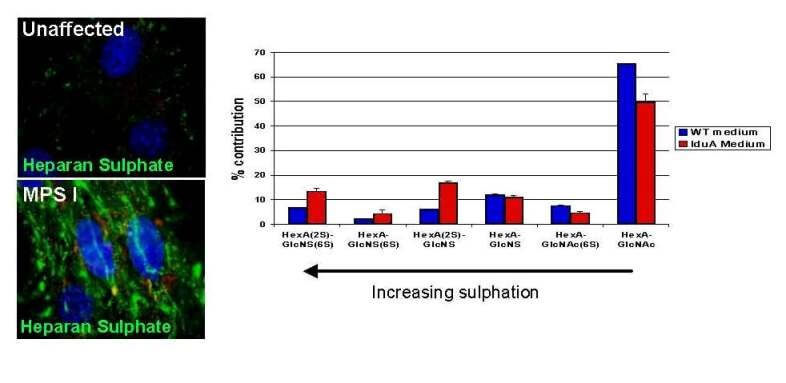
We demonstrated that MPS I bone marrow cells do not only store HS inside the cells, butoutside cells in the extracellular matrix (ECM). The ECM is a sticky material made up of proteins and sugars which anchors and displays chemokines for interaction with homing cells. In the photographs (above left), heparan sulphate is stained fluorescent green, and there is clearly more associated with MPS I cells (bottom image). In the graph (above right), each bar represents the percentage of disaccharide building blocks that have sulphate groups. MPS I bone marrow has a dramatic increase in sulphated disaccharides in HS. This shows that MPS I HS is much more highly sulphated than normal. The amount of sulphation is important in chemokine-HS interactions.
Year 2: High Levels of Highly Sulphated Heparan Sulphate are Inhibitory to Stem Cell Homing
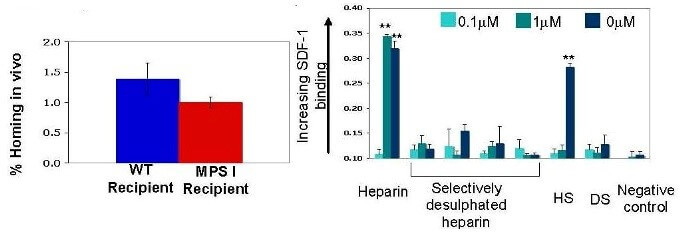
Experiments in vivo showed decreased homing in MPS I recipients of bone marrow transplants (above left). Work in vitro showed that heparan sulphate decreased the ability of bone marrow stem cells to home to the chemokine SDF-1, which is the most important chemokine for bone marrow transplant. This work showed that at low concentrations, the inhibition of homing was not dependent on the sulphation state of HS, however at high concentrations, sulphation state became important. A binding experiment (above right) showed that this was because SDF-1 will only bind to highly sulphated forms of HS at high concentrations it does not bind to desulphated forms at all.
Conclusions
We concluded that the decreased homing seen in transplants to MPS I recipients was caused by the unique combination of HS that is both highly sulphated and in massive excess in MPS I bone marrow cells, which causes SDF-1 to become trapped in super-sticky ECM, preventing it from signalling to transplanted cells. This is a disease-specific defect in homing that could be contributing to graft failure in the clinic.
Awards
This work was presented at the Harden Conference on Heparan Sulphate in Cambridge in 2009 , where it was awarded the Mituzani Foundation Poster Prize. It was also selected for oral presentation at the Clinical and Laboratory Sciences Showcase, University of Manchester 2009.
Dr. Adriana M Montano
Saint Louis University School of Medicine
St Louis, MO
Identification of genes for keratin sulfate biosynthesis: toward development of RNAi mediated therapy
Study has extended to 2011
Dr. Mark S. Sands
Washington University School of Medicine
St. Louis, MO
Goals:
1) determine the effects of reduced lysosomal recycling on the energy imbalance in affected cells, and
2) determine the effects of dietary intervention on the progression of disease.
We previously showed that there was a significant energy imbalance in the form of depleted adipose stores in mouse models with lysosomal storage diseases (three of which were MPS disorders). We hypothesized that the energy imbalance was due to interrupted lysosomal recycling of macromolecules and the subsequent increase in energy expenditure required for the synthesis of new macromolecules. In order to further test this hypothesis we measured the levels of over 1,500 metabolites in liver homogenates from MPS I mice. Molecules directly involved in energy utilization such as simple sugars, nucleic acids and lipids were depleted. This is consistent with the animals being in a state of nutrient depravation. Interestingly, the levels of amino acids, amino acid derivatives and dipeptides were increased suggesting that protein catabolism, perhaps due to increased autophagy, is at least partially fulfilling intermediary metabolism. We showed that autophagy is increased in the livers of both MPS I and VII mice. The initial observation of energy imbalance also led us to hypothesize that a diet rich in energy (simple sugars and lipids) would decrease the severity of the disease. Therefore, we put both MPS I and VII mice on a high fat, simple sugar diet. Although there was no significant increase in life span or retinal function, most of the abnormalities identified on the metabolomic screen approached normal levels. In addition, the animals were now able to increase their adipose stores and autophagy was reduced to near normal levels. These data support our hypotheses that interrupted lysosomal recycling leads to a significant energy imbalance and that nutritional intervention can ameliorate some of the biochemical abnormalities associated with these diseases. This work was recently published [Woloszynek JC, et al., (2009) J. Biol. Chem. 284:29684-29691].
This grant also partially supported a study in which we combined adeno-associated virus (AAV)-mediated, CNS-directed gene therapy with bone marrow transplantation (BMT) in the murine model of MPS IIIB. We hypothesized that systemic therapy (BMT) would synergize with CNS-directed gene therapy to increase efficacy in MPS IIIB which has been relatively refractory to most therapies. CNS-directed, AAV-mediated gene therapy alone provided the greatest clinical benefit. The addition of BMT added little in the way of efficacy and actually decreased the life span slightly. This study was recently published [Heldermon C, et al. (2010) Mol. Ther. 18:873-880]. We have initiated another study in the MPS IIIB mouse where we combined CNS-directed AAV-mediated gene therapy with systemic lentiviral-mediated gene therapy. The rationale for this study is that the systemic lentiviral-mediated gene therapy would result in persistent high levels of circulating NaGlu activity that will enhance the effects of the CNS-directed therapy. It has been shown in another model of MPS that persistent high levels of circulating enzyme can result in some CNS correction. The preliminary data (14-16 months of age) suggest that the combination of AAV-mediated CNS-directed gene therapy combined with systemic gene therapy provides the greatest clinical benefit.
Publications:
Woloszynek JC, Kovacs A, Ohlemiller KK, Roberts M, Sands MS: Metabolic adaptations to interrupted glycosaminoglycan recycling. J Biol Chem, 284:29684, 2009.
Heldermon C, Ohlemiller KK, Herzog E, Vogler C, Qin E, Wozniak DF, Tan Y, Orrock J, Sands MS: Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy or both in the mouse model of MPS IIIB. Mol. Ther., 18:873, 2010.
Dr. Marta Serafini
Dulbecco Telethon Institute at M.Tettamanti Research Center Clinica Pediatrica Univ.
Monza, Italy
Marrow mesenchymal stem cell therapy for MPS I
The overall aim of ongoing experiments on this project is to establish a new stem cell-based therapy to improve the outcome of hematopoietic cell transplantation with a particular emphasis on the skeletal defects affecting MPS-I patients. For this purpose, we have used two complementary cell systems: on one side human mesenchymal stem cells (hMSCs) and on the other murine mesenchymal stem cells (mMSCs).
We have successfully isolated hMSCs from healthy donors and three MPS-I patients. We have evaluated their intrinsic osteogenic differentiative potential in vitro and, so far, our data indicate that genetically affected cells do not differ from healthy donors-derived cells, suggesting that microenvironment and/or signalling from neighbouring cells might be responsible for the observed phenotype at skeletal level. In order to investigate the best source of stem cells, hMSCs have been isolated from healthy donors also from umbilical-cord blood, amniotic fluid and umbilical cord with an increasing efficiency in our system (around 70% for bone marrow and amniotic fluid, around 80% for digested umbilical cord and around 25% for umbilical cord blood).
These cells differentiate in vitro in adipocytes, chondrocytes and osteoblasts and we are now evaluating their in vivo potential. In parallel, we sought to isolate and characterise mMSCs. So far two protocols have been used and tested as it is widely accepted that mMSCs isolation protocols are less established. With our methods by the first passages we obtain a mixed population with an average of 50% of MSCs assessed by detection of markers as CD73, CD29, CD44, SCA1, CD36 and negativity for B220, CD11b and CD45 (by antibody staining), and good potential to differentiate in adipocytes, osteoblasts and chondrocytes. We are now in the process of isolating and culturing mMSCs using an inactivated feeder layer of murine fibroblasts, as a mean to increase the homogeneity and efficiency of mMSCs isolation. In order to test the rescue capability of HCT with or without mesenchymal stem cells (MSCs), we planned to use the mouse model as our experimental system. To date, different murine models have been reported with mutations in the IDUA gene. Originally we established to use an immunodeficient mouse, the NOD/SCID/MPS-I (Garcia-Rivera et al., 2007) infused with human-derived MSCs.
Before starting the transplant experiments, we analysed the skeletal phenotype of this and other mutant models and we found that the range of bone-defects varied in penetrance and severity across the mutants. This situation could be compared to the differences observed in phenotype in MPS-I patients, representing a valuable tool to study the feasibility of MSCs transplant also in less severe forms of MPS-I.
In the last few months we have isolated GFP+ mMSCs and transplanted them into the different MPS-I mouse models. At defined time points after the infusion, we plan to evaluate the skeletal phenotype improvement, with or without hematopoietic stem cells. We have also tested several injection sites as MSCs are technically challenging to infuse for their intrinsic tendency to adhere. As demonstrated by our and other groups, intravenous injections are often lethal to the recipient animal, and cells tend to colonise the lungs and the heart but cells are rarely detectable in other inner organs and/or bones. We are now evaluating in site injections, intra-cardiac injections and possibly the use of cells containing-scaffold materials.
Dr. Richard Steet
University of Georgia Research Foundation
Athens, GA
Investigation of the cartilage pathogenesis of ML II and MPS
Defining the pathogenic mechanisms of MPS and MPS-related disorders is important since it will potentially aid in the development of new therapies. Our proposed research was designed to take advantage of the power and speed of the zebrafish system to identify factors that contribute to the cartilage pathogenesis in mucolipidosis II or ML-II, with the goal of directly investigating the contribution of these factors towards the disease process. With ongoing support from the MPS Society, we have now identified and confirmed that several enzymes – including cathepsins K, L and S and matrix metalloproteinase 13 – are upregulated in ML-II zebrafish embryos as well as feline ML-II tissues (Petrey A., Flanagan-Steet H., Nairn A., Moremen K., Haskins, M. and Steet R., manuscript in preparation). The activity of the cathepsins is elevated in ML-II zebrafish at time periods when only marginal activity can be detected in wild type embryos, suggesting that sustained or unregulated expression of these hydrolases occurs in response to impaired mannose phosphorylation.
This abnormal expression may also compound the hypersecretion of these enzymes caused by defective mannose 6-phosphate dependent lysosomal targeting. Our current studies are focused on 1) localizing the increased cathepsin expression within specific tissues of our zebrafish model and 2) testing whether a reduction in this activity by genetic or pharmacological means will have therapeutic potential. In the coming years, we plan to translate any positive outcomes from our zebrafish experiments to studies in the feline ML-II model in collaboration with Dr. Haskins at the University of Pennsylvania. Another facet of our MPS Society-funded project was to generate models for other MPS disorders using the zebrafish system, with the hope of using these models to explore whether common pathogenic mechanisms exist between ML-II and MPS disorders. Although we were able to reduce the activity of beta-glucuronidase (GUSB; the cause of MPS VII), in zebrafish embryos to less than 5% of wild type levels, no obvious phenotypes were observed. These data suggest that the relatively short duration of morpholino-induced gene suppression may limit our ability to model the more progressive MPS disorders in this organism. This finding led us to seek new ways to establish stable genetic zebrafish mutants with sustained loss of enzyme activity. In a fortunate respond to our request, GUSB was selected by an NIH-funded initiative that is tasked with generating zinc-finger nuclease constructs suitable for the stable suppression of genes in zebrafish. The development of a zebrafish model for MPS VII will be ongoing in the second half of this year in our lab and will expand the experimental systems available to investigate MPS pathogenesis.
As a new investigator, I am truly grateful to the MPS Society and the families for their support of this research project. Funding from the Society has proven vital in our ability to pursue higher risk experiments and we remain hopeful that the avenues of research that stem from this work can be translated into therapies for MPS and MPS-related disorders.
2007 Research Grants
The National MPS Society has awarded $508,000 in new grants for 2007. Drs. Biffi, Haskins and Simonaro were awarded the general research grants of $100,000 total. Dr. Fuller was awarded the $90,000 MPS II grant, and Dr. Mellon was awarded the MPS III grant for $80,000. Each grant is for two years, and the researchers will receive half of the total each year.
This year we collaborated with two foundations to offer partnership grants. In May, in partnership with the Ryan Foundation, we awarded $19,000 to Drs. Katherine Ponder and Mark Haskins for their work in “Retroviral vector-mediated gene therapy for MPS I”. The second partnership grant of $19,000 will be offered in conjunction with ISMRD (International Society for Mannosidosis and Related Diseases). We will report the details of that grant in our next newsletter.
Drs. Katherine Ponder and Mark Haskins Partnership Grant with the Ryan Foundation “Retroviral vector-mediated gene therapy for MPS I” Washington University School of Medicine (Dr. Ponder) St. Louis. MO
University of Pennsylvania, School of Veterinary Medicine (Dr. Haskins) Philadelphia, PA
Mucopolysaccharidosis I (MPS I) is a lysosomal storage disease caused by deficient α-L- iduronidase (IDUA) activity, which results in the accumulation of the glycosaminoglycans heparan and dermatan sulfate. The severe form, known as Hurler syndrome, causes bone and joint abnormalities, pulmonary and cardiac disease, hearing and visual deficiencies, mental retardation, and death around age 5 if untreated. Hematopoietic stem cell transplantation can reduce some manifestations, but has a 15% mortality rate, costs $130,000, and requires a compatible donor. Enzyme replacement therapy can also reduce some symptoms, but costs over $500,000 per year for an adult, requires a weekly infusion, and is not available to all patients. The development of an effective and safe gene therapy for MPS I could have a dramatic positive impact on the lives of patients and the families that care for them. We previously demonstrated that neonatal intravenous injection of a gamma retroviral vector (γ-RV) with an intact long- terminal repeat (LTR) expressing canine IDUA had a truly remarkable effect in both mice and dogs with MPS I, with elimination or reduction in all major clinical manifestations. This was due at least in part to efficient transduction of liver cells, which secreted mannose 6-phosphate (M6P)-modified IDUA into blood, which diffused to other organs and was taken up via the M6P receptor. There was also some transduction of blood cells and an undefined cell type in brain, which may have contributed to the therapeutic response. Although no tumors developed in mice or dogs with this approach, the risk of insertional mutagenesis with an LTR-intact vector is a concern. Another problem is that administration of this vector to adult MPS I mice or newborn MPS I cats resulted in a potent cytotoxic T lymphocyte (CTL) response that destroyed transduced cells. The aims of this project are to: 1) reduce the risk of insertional mutagenesis by developing a self-inactivating γ-RV with a deletion in the enhancer of the 3′ LTR; 2) attempt to prevent an immune response by avoiding expression in antigen-presenting cells; and 3) analyze the duration of efficacy and evaluate for toxicity in a long-lived large animal model (dog). If successful, this study may hasten the development of a simple and effective treatment for newborn patients that will reduce or prevent the devastating clinical manifestations of MPS I.
“Novel efficacious and safe gene therapy approaches for the treatment of MPS I”
San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) Milano, Italy
Type I Mucopolysaccharidoses (MPSI) is a lysosomal storage disorder (LSD) due to the inherited deficiency of a-L-iduronidase (IDUA) and the resulting accumulation of its toxic substrates in many organs. Among MPSI clinical variants, the Hurler syndrome is fatal in childhood, and represents the form with the higher need for the development of new efficacious therapies, capable of alleviating all disease-related symptoms. Indeed, despite several experimental therapies have been tested both in MPSI animal models and in patients, no efficacious treatment is currently available for the cure of Hurler syndrome. This lack of efficacy is likely due to the difficulty of providing sufficient amount of the functional IDUA to all disease sites, including the brain, in the absence of toxicity. Therefore, the main goal of the project is the identification of a novel gene therapy strategy capable of efficiently deliver therapeutic levels of functional IDUA enzyme to all disease sites of MPS I mice, and of correcting disease manifestations, in the absence of toxicity. To this goal, based on our expertise in other LSD models, we will compare two gene therapy protocols based on advanced generation viral vectors, which might over-come the major limitations of currently available therapies. This work will allow us to identify and further develop towards clinical application the most promising and efficacious gene therapy strategy for the treatment of MPSI.
“Lentiviral Vector Therapy for Canine MPS VII” University of Pennsylvania, School of Veterinary Medicine Philadelphia, PA
Based upon experiments in mice, a clinical trial has been approved for our collaborator, Mark Sands, PhD, to use a lentiviral vector containing the human gene for the enzyme that is deficient in mucopolysaccharidosis VII to treat bone marrow cells in culture and then return them to the children with MPS VII. Currently, the clinical trial is on hold while Dr. Sands collects more safety data for the FDA. We have a well-characterized dog model of MPS VII and believe it is essential to test the safety and efficacy of this therapy in MPS VII dogs prior to its use in children. We also have successfully treated MPS VII dogs intravenously with a retrovirus vector at three days of age dramatically improving the skeletal, ocular, and cardiac lesions. Five treated dogs are currently more than 6 years post-treatment and are being maintained to evaluate possible long-term side effects of therapy, together with four dogs treated by intravenous, neonatal adeno-associated virus vector gene therapy.
“Pathogenesis and Treatment of the Mucopolysaccharidoses”
Mount Sinai School of Medicine New York, NY
The past decade has witnessed remarkable advances in the understanding and treatment of the MPS. However, despite these advances, major challenges remain. For example, although enzyme replacement therapy (ERT) has recently become available for several of these disorders, it is extremely expensive and requires life-long infusions of recombinant enzyme. ERT also has very limited effects on the bones and joints, major sites of disease in MPS patients. Our laboratory has been using MPS VI animal models to study the mechanism of disease in bones and joints, as well as to evaluate new approaches to treatment. This research has led to a better understanding of the specific changes that occur in these tissues, facilitating the future design of more effective therapies. In the current proposal we will extend these findings and pursue three aims. In the first we will continue to investigate the mechanism by which GAG storage leads to bone and cartilage destruction using cells from MPS VI rats. In the second we will obtain fluid from the joints of MPS VI cats, and measure the levels of several proteins to see if they are abnormally expressed. We will determine the level of these proteins as a function of age, and evaluate whether they can be used to predict the severity of disease and/or the outcome of treatment in the bones and joints (i.e., biomarkers). In the last aim we will use MPS VI rats to evaluate the effects of two clinically available “anti-inflammatory” medications on the progression of disease, as well as one experimental medication that targets a pathway we have found abnormal in MPS VI cells. If we obtain evidence in the rats that such therapies are effective, in the future these approaches could be evaluated in MPS patients, alone or as adjuncts to ERT.
“Membrane microdomains and improved clinical management for the mucopolysaccharidosis”
Children, Youth and Women�™s Health Service North Adelaide, SA, Australia
The mucopolysaccharidoses (MPS) are chronic progressive genetic diseases that generally affect young children. Symptoms are debilitating and progressive, and include heart and breathing difficulties, skeletal deformity and brain degeneration. The MPS result from the progressive storage of waste in a component of each cell known as the lysosome. In affected children, the accumulation of this waste interferes with each cell�™s normal functioning and leads to the deterioration and death of cells, organs and tissues. There are no cures for MPS and current treatment options are not without their limitations. Although the underlying genetic defects have been determined for many MPS, the disease process remains poorly understood. The diverse array of clinical symptoms in MPS suggests that many cellular processes are altered. A major one is likely to be the fat composition and distribution in cells. Fats have been shown to be altered in the MPS and this project proposes to examine the types of fats that are altered and their location in the cell. Once we understand the changes in fats, we will attempt to correct these changes using conventional drugs and fatty acid manipulation. Successful studies performed in cells in this project will pave the way for further studies in animal models to see if the pathology in MPS can be treated with diet and drugs.
“Neurosteroid treatment of MPS IIIA”
University of California, San Francisco
Department of Obstetrics, Gynecology & Reproductive Sciences San Francisco, CA
We have identified a potential treatment for a lysosomal storage disorder that involves a class of biological compounds called neurosteroids. These compounds are synthesized in the brain in a developmentally programmed fashion. Among their many effects, they have effects on development of new neurons, survival of neurons, protection against toxicity to neurons. We showed that treatment of a mouse model of the lysosomal storage disorder Niemann Pick Type C (NP-C) with the neurosteroid allopregnanolone doubles lifespan, delays loss of motor function, and rescues neurons that die in NP-C. We now have preliminary data in MPS IIIA mice that a similar treatment with allopregnanolone will enhance lifespan, delay loss of motor function, increase muscle strength, and reduce aggressive behavior. We now propose to expand these studies to include more mice to assess the effect of allopregnanolone treatment on MPS IIIA 1. longevity 2. locomotor function 3. neuronal survival 4. neuronal storage and 5. begin to assess peripheral markers of disease progression and effective allopregnanolone treatment. Successful completion of these aims should provide preliminary data for submission of a larger grant to the NIH.
Mt. Sinai School of Medicine, New York, NY
“Pathogenesis and Treatment of the Mucopolysaccharidoses
Received 7-08
The underlying premise of our research is that despite some successes, new treatment strategies are needed to replace, or more likely augment, existing approaches for the MPS disorders. This is particularly true for the bones and joints. In order to develop such approaches, as well as to identify new biomarkers for these diseases, a better understanding of the disease mechanism at the sites of pathology is necessary. Due to the limits of human experimentation, this can best be accomplished using animal model systems.
Based on our preliminary findings, we have chosen to evaluate therapeutic strategies that slow or prevent the pro-apoptotic and pro-inflammatory effects of GAG storage (see below). We have shown that inflammation is a major component of the MPS disorders, although to date the effects of anti-inflammatory medications on these diseases have not been systematically evaluated.
During the past year we have further characterized the inflammatory disease that occurs in MPS VI and other MPS animal models. In addition, we have identified many biomarkers that can be used to monitor disease progression and treatment. One new paper describing these findings has been published this past funding period (Simonaro et al. Am. J. Path. 172, 2008), which represents the third in a series describing the mechanism of joint disease in MPS.
San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET)
Milano, Italy
“Novel efficacious and safe gene therapy approaches for the treatment of MPS I�?
Received 7-08
The pipeline of the proposed project consisted in the comparison of the long-term efficacy in alleviating disease manifestations in the MPSI mouse model of three different therapeutic approaches:
To this aim, during the 1st year of funding we focused on:
The MPSI colony was fully established in our animal facility. We applied different relevant outcome measures to MPSI affected homozygous, as well as to unaffected litters, including:
This preliminary evaluation was fundamental for defining proper readouts for our long-term efficacy studies.
We constructed and produced LV encoding the human IDUA under the control of the human PGK promoter, with the addition of the W-PRE element to increase transgene expression (PGK.IDUA.LV). This vector was used to transduced human (CD34+, from normal cord blood) and murine (lineage negative selection from the bone marrow of both wild type and MPSI donors) HSC, according to already established protocols. Transduction allowed sustained IDUA expression at supra-physiological levels (up to 20 fold above basal levels). IDUA over- expressing cells retained a normal proliferation and differentiation capacity al clonogenic assays in vitro. Further, upon transplantation into adequate models (irradiated MPSI mice for murine HSC, and immunodeficient Rag2-/- gamma chain -/- mice for human HSC), IDUA over- expressing cells demonstrated long-term engraftment and repopulation potential. Overall, these data demonstrate the feasibility and safety of LV-mediated IDUA over-expression in HSC.
HSC were isolated by lineage negative selection from 6-8 weeks old wild type littermates and transduced with control LV encoding GFP under the control of the human PGK promoter, and then transplanted into lethally conditioned homozygous defective MPSI mice, at 2 months of age, to determine the possibility to achieve prevention/correction of disease manifestations. As we already demonstrated that the extent of tissue macrophages and microglia replacement is not affected by the conditioning regimen applied prior to the transplantation, we lethally irradiated recipient mice. The engraftment of donor-derived, GFP transduced cells measured by FACS analysis on peripheral blood mononuclear cells was above 70% in all transplanted animals. A cohort of 15 transplanted animals is currently available for efficacy evaluations. Efficacy of the treatment will be evaluated on 10 months old treated mice, looking at prevention and correction of functional, biochemical and histopathological abnormalities.
Group B: HSC gene therapy with IDUA LV
We transplanted HSC from homozygous defective MPSI mice, transduced with PGK.IDUA.LV, into lethally conditioned, 2 months old IDUA-/- mice. IDUA expression levels measured on the in vitro progeny of transduced HSC by 4-methylumbelliferyl assay showed sustained IDUA over-expression (above20 fold the basal levels) and vector content was quantified by LV-specific quantitative PCR analysis (5-6 LV copies/genome). A cohort of 20 transplanted animals is currently available for efficacy evaluations. IDUA specific activity will be measured on cell lysates from PBMC, starting from 8 weeks after the transplant. Efficacy of the treatment will be evaluated on 10 months old treated mice showing IDUA activity reconstitution above wild type levels in PBMC, looking at prevention and correction of functional, biochemical and histopathological abnormalities. Comparison with the outcome of allogeneic HSCT (group A) and with MPSI mice transplanted GFP transduced IDUA-/- HSC will be performed, in order to assess the therapeutic role of enzyme over-expression in the HSC progeny.
Group C: Liver-directed gene therapy with miRNA regulated LV
We constructed and produced LV encoding the IDUA cDNA under the ET or the PGK promoters with and without the addition of target sequences for the mir142-3p, which is robustly expressed in hematopoietic cells, such as. During the next few months, two months old homozygous defective MPSI mice will be injected intravenously with 5×108 IU of the tagged LV.ET.IDUA.142-3pT or control LV.ET.GFP.142-3p. ELISA will monitor appearance of neutralizing anti-IDUA or anti-GFP antibodies starting from 7 days after vector administration. Treated animals lacking evidence of antibody production and controls will be evaluated at 10 months of age for prevention and correction of functional, biochemical and histopathological abnormalities and for vector content in affected organs by LV-specific quantitative PCR.
School of Veterinary Medicine, University of Pennsylvania Philadelphia, PA
“Lentiviral Vector Therapy for MPS VII�?
Received 7-08
Our initial experimental plan was based upon experiments in mice, from which a clinical trial had been approved for our collaborator, Dr. Mark Sands, to use a lentiviral vector containing the human gene to treat MPS VII bone marrow cells in culture and then return them to the children with MPS VII. Over the past year, we transplanted seven dogs with gene modified autologous bone marrow. Three control dogs received cells expressing the marker GFP and four MPS VII dogs received cells expressing beta-glucuronidase (GUSB). Briefly, stem cells were isolated from bone marrow aspirates and exposed to recombinant lentivirus supplied by Dr. Sands either overnight or in two cycles over 48 hours. The transductions took place in serum free conditions and the cells were supplemented with cytokines. For the control dogs, 9.7+/-2.7% of the cells were determined to be GFP positive by flow cytometry and 0.18+/-0.08 x 106 cells/kg were returned to the donor dogs. The MPS VII dogs received 1.51+/-1.27 x 106 cells/kg, and 30.1+/- 3.3% of those cells expressed GUSB histochemically. No significant number of GFP positive cells was detected in peripheral blood from the control dogs. For the MPS VII dogs, where flow was unavailable, no increase in GUSB activity was detected in peripheral blood cells or in serum. However, vector derived sequence was detected in two of the dogs by PCR. For one dog there was a single positive time point two weeks after the transplant while the second dog remained positive for the duration of the study. Because of the difficulty in translating this technique from the mouse to the dog, the study was put on hold. A vital aspect of the Society’s funding was to help support the colony of MPS VII dogs and, in particular, those that had been successfully treated intravenously as neonates using a retrovirus vector. These dogs were 6 years old and had dramatic improvement in the skeletal, ocular, and cardiac lesions but were at risk during a funding lapse from the NIH. The five treated dogs are now 7.5 years post-treatment and are being maintained to evaluate continued efficacy and possible long-term side effects of therapy. We presented a talk “Seven-Year Update for Neonatal Intravenous Retroviral Treatment of MPS VII Dogs�? at the American Society for Gene Therapy in Boston. In addition, studies on MPS VII dog’s bones and joints over the past year have resulted in two papers (Simonaro, C., D’Angelo, M., et. al. (2008) Mechanisms of glycosaminoglycan-mediated disease: Implications for the mucopolysaccharidoses and other connective tissue diseases. Am J Pathol 172:112-122, and Herati, R.S., Knox, V.W., et. al. (2008) Radiographic evaluation of bones and joints in mucopolysaccharidosis I and VII dogs after neonatal gene therapy. Molec Genet Metab, in press). We have also evaluated the effects of high and low serum GUSB activity resulting from retroviral gene therapy on the lesions in the brain and our preliminary conclusion, presented at the 10th International MPS and Related Disease Conference in Vancover, is that high circulating enzyme appears to reduce GAG storage in the hippocampus. In addition, we have now treated 10 MPS VII dogs intravenous as either neonates or at 45 days of age with adeno-associated viral (AAV) vectors of different serotypes. These dogs are not doing as well clinically as those treated with the retroviral vector, but are being maintained to evaluate longer-term therapy. Future studies. 1) We have been successful in achieving prevention of many aspects of MPS VII using a retroviral vector. However, because of concerns about insertional mutagenesis and the risk of cancer, we have developed a self-inactivating retrovirus vector that should add a layer safety to the treatment. Over the next year, we will make vector virus and administer it to MPS VII dogs to evaluate efficacy and long-term safety. 2) We will continue to evaluate the MPS VII dogs treated with AAV vectors. 3) We have now established a collaboration with the Penn Center for Musculoskeletal Disorders to evaluate the physical properties of ligaments, tendons, and articular cartilage in the joints of dogs with MPS VII. These data, combined with those of Lilla Simonaro and Kathy Ponder on the biochemistry of the structures, will be used to devise a pharmacological approach to therapy of the joints.
Children, Youth and Women’s Health Service North Adelaide SA, Australia
“Membrane microdomains and improved clinical management for the mucopolysaccharidoses�?
Received 9-08
The mucopolysaccharidoses (MPS) are characterised by the lysosomal accumulation of sugars, which is the primary cause of disease. However, the diverse and extensive array of clinical symptoms in MPS disorders suggest that many other cellular processes and functions are involved in the onset of symptoms and the rate at which they progress. One such process is likely to be the composition of certain fats and their distribution in cells. Within cells, specialised regions [or domains] of fat exist, which are known as rafts. Rafts are composed of certain types and structures of fats and they play important roles in cell communication and coordination; they are crucial for cell function. If the fat composition of the rafts is altered, it is likely that they cannot function properly and this may be a mechanism leading to disease in MPS. With the funding provided by the National MPS Society, we hope to better understand the changes in these fats and attempt to correct these changes to see if we can bring them back to normal using conventional drugs and fatty acid supplementation. For the purpose of this study, we are using cultured MPS skin cells as cell models of the MPS disorders.
Aim 1: Determine the fat composition of specialised domains
In the laboratory environment, rafts can be isolated from other fats in the cell by their resistance to solubilisation by detergent, i.e. they float while the other fats are absorbed by detergent. However, in practice, isolating rafts from skin cells has proved to be problematic; the number of cells used in raft preparations and the growth rate have been shown to be important factors. Overcoming these particular problems has impeded our progress. We found that it was not possible to isolate rafts from slow growing cells and that 2 mg of cell protein was required. None of our unaffected control cell lines in the archives were suitable so we needed to collect fresh skin biopsies from healthy volunteers for this purpose. These cells are now growing well and raft isolation is underway. In the meantime, however, we have successfully isolated rafts from three MPS I cell lines and their fat composition has been determined by a sophisticated technique known as mass spectrometry. Once the rafts have been isolated from the control cell lines and have been fully analysed we will compare the differences between them and the MPS I cells. We will also then isolate rafts and determine the fat composition in MPS II, IIIA and VI cells.
Aim 2: Modulate the fat composition of the specialised domains back to normal
Even though we have not yet determined which fats are altered in MPS compared with unaffected controls, we have performed some preliminary experiments to demonstrate that we could alter the structure and types of fats within the MPS cells. Firstly, we inhibited the production of one of the fats by the addition of the drug myriocin to the culture media in which the skin cells are being grown. This was successful and reduced the amount of this fat in the cells by 50%. Next, to demonstrate that we can modify the structures of the fats in MPS cells we added fatty acids to the culture media. We showed that the addition of linoleic and oleic acids to the culture media was able to alter the structures of some of the fats present. Now that we have overcome the problems of raft isolation we will be able to complete Aim 1, and then we can identify which fats are altered and attempt to correct them in this aim.
Aim 3: Evaluate fats as biomarkers of disease in MPS
We have identified some potential fats that may be useful as biomarkers. Future work will involve measuring them in cells from MPS patients of different genotype and phenotype to evaluate their usefulness.
Dept. of OB, GYN, Univ. of Calif. San Francisco San Francisco, CA
“Neurosteroid treatment of MPS IIIA�?
Review not received; funding not provided for second year
2nd Year Research Reviews
“Novel efficacious and safe gene therapy approaches for the treatment of MPS I�?
San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET)
Milano, Italy
received 8-09
The pipeline of the proposed project consisted in the comparison of the long-term efficacy in alleviating disease manifestations in the MPSI mouse model of three different therapeutic approaches:
To this aim, during the 2nd year of funding we focused on:
immunodeficient Rag2-/- gamma chain -/- mice. The novel strain reproduces the main pathologic features of MPS1 and retains the key properties of Rag2-/- gamma chain -/- mice. Indeed, these animals allow transplantation, engraftment and differentiation of human HSC. Therefore, they are currently been employed for assessing the therapeutic potential of human HSC (either from normal donors or MPS1 patients, upon gene correction with IDUA-encoding LV) in correcting MPS1 disease manifestations.
The efficacy of HSC-based gene therapy has been evaluated tested in IDUA KO mice (C57Bl6 background), which were transplanted at 2 months of age with either wipe HSC (transduced with GFP-encoding LV) or HSC from MPS1 donors, transduced with LV encoding the human IDUA. At the age of 8 months, reconstitution of enzymatic activity up to 100 fold above the levels detected in wild type mice was observed in peripheral blood mononuclear cells of mice receiving the gene corrected cells, whereas restoration of normal IDUA expression levels was seen in mice receiving the wild type HSC. At this time-point the phenotype of treated mice was evaluated by the means of functional, biochemical and histopathological studies. Wild type HSC transplantation allowed reaching physiological values of enzyme activity in the liver, spleen and kidney, but was unable to reconstitute detectable levels of IDUA activity in the hearth and brain of treated animals Importantly, gene therapy led to enzyme over-expression in liver and spleen (to comparable levels with the hematopoietic system), and to reconstitution of enzyme activity in the kidney, hearth and brain at least up to the levels detected in the corresponding organs from wild type mice. Moreover, a rescue of major phenotypic abnormalities was observed in gene therapy treated mice as compared to controls. Gene therapy
treated animals showed amelioration of both the skeletal abnormalities and the behavioral performances as compared not only to mock-treated MPS1 controls, but also to MPS1 mice receiving wild type HSC. Histopathological evaluation of the tissues collected from all the experimental groups is currently on going.
We would like to thank you for this funding opportunity and look forward collaborating with you again.
School of Veterinary Medicine, University of Pennsylvania Philadelphia, PA
“Lentiviral Vector Therapy for MPS VII�?
Received 8-09
As reported last year, our initial experimental plan was based upon experiments in mice by our collaborator, Dr. Mark Sands, to use a lentiviral vector containing the human gene to treat MPS VII bone marrow cells in culture. In the previous year, we determined there were difficulties in translating this technique from the mouse to the dog and the study was put on hold.
A vital aspect of the Society’s funding was to help support the colony of MPS VII dogs and, in particular, those that had been successfully treated intravenously as neonates using a retrovirus vector. We have now had to euthanize two of the dogs, one with a hematoma of the spleen and one following an infection in her front leg. Both dogs had lived for ~8 years and were ambulatory, although with advancing age they and the other two dogs who are still alive in the colony find getting around more difficult. The dogs continued to have clear corneas and competent mitral valves.
In addition, we have now treated 10 MPS VII dogs intravenous as either neonates or at 45 days of age with adeno-associated viral (AAV) vectors of different serotypes. These dogs have not done as well clinically as those treated with the retroviral vector, but were maintained to evaluate longer-term therapy.
Future studies. 1) We have been successful in achieving prevention of many aspects of MPS VII using a retroviral vector. However, because of concerns about insertional mutagenesis and the risk of cancer, Dr. Kathy Ponder developed a self-inactivating retrovirus vector that should add a layer safety to the treatment. Over the next year, we will make vector virus and administer it to MPS VII dogs to evaluate efficacy and long-term safety. 2) We will continue to evaluate the MPS VII dogs treated with AAV vectors. 3) We have now established a collaboration with the Penn Center for Musculoskeletal Disorders to evaluate the physical properties of ligaments, tendons, and articular cartilage in the joints of dogs with MPS VII.
These data, combined with those of Drs. Lilla Simonaro and Kathy Ponder on the biochemistry of the structures, will be used to devise a pharmacological approach to therapy of the joints.
The funding by the MPS Society was critical to be able to keep the research on track during a lapse from the NIH. Thank you.
Mt. Sinai School of Medicine, New York, NY
“Pathogenesis and Treatment of the Mucopolysaccharidoses�?
Received 7-09
Therapies are available for some MPS disorders with limited effects in the bones and joints. Therefore, the overall goal of our research has been to use MPS animal models to study the disease mechanism in these tissues in order to develop new and improved therapeutic approaches. We have previously established that inflammation plays a major role in the pathology of MPS bones and joints, and that prevention of inflammation may have an important therapeutic effect. Our recent generation of an MPS mouse model with an inactivated inflammatory pathway (see below) has proven that inflammation has an important role in the pathogenesis of MPS bone disease. During the past year we have also continued long-term studies using the FDA-approved anti-inflammatory drug, Remicade™, in MPS VI rats. We are hopeful that completion of the Remicade™ studies in the MPS VI rats will provide a basis for the initiation of clinical trials, and “fast-track�? approval of this (and perhaps other anti- inflammatory) drugs for MPS patients.
Figure 1.
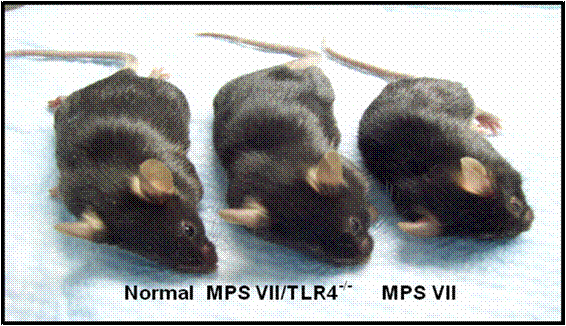
The photo above is representative of a five-month olf normal male,. MPS VII/TLR4 and MPS VII mouse.
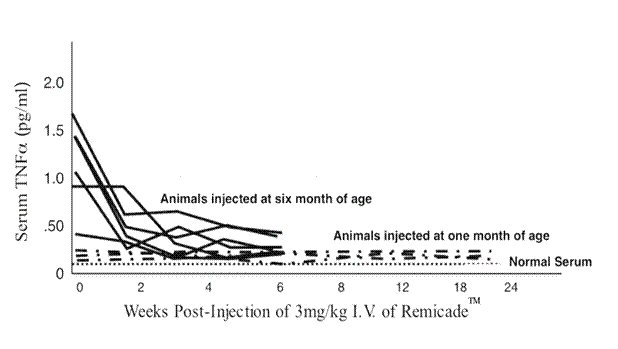
Dr Maria Fuller
Children, Youth and Women’s Health Service North Adelaide SA, Australia
“Membrane microdomains and improved clinical management for the mucopolysaccharidoses”
received 9-09
The mucopolysaccharidoses (MPS) are characterised by the lysosomal accumulation of glycosaminoglycans, which is the primary cause of disease. However, the diverse and extensive array of clinical symptoms in MPS disorders suggest that many other cellular processes and functions are involved in the onset of symptoms and the rate at which they progress. One such process is likely to be the composition of certain lipids and their distribution in cells. Within cells, specialised regions [or membrane microdomains] of lipid exist, which are known as rafts. Rafts are composed of certain types and structures of lipids and they play important roles in cell communication and coordination; they are crucial for cell function. If the lipid composition of the rafts is altered, it is likely that they cannot function properly and this may be a mechanism leading to disease in MPS. With the funding provided by the National MPS Society, we have developed methodology to isolate rafts from cultured MPS skin cells (fibroblasts), which we are using as models of MPS disease. Furthermore, using a mass spectrometry “lipidomic�? approach we have shown that the lipid composition of these raft domains in the MPS cells is different to that of the control cells.
In the first year of the project we developed methodology that would allow us to isolate lipid rafts from cultured skin fibroblasts reproducibly. We found that it was not possible to isolate rafts from slow growing cells and that 2 mg of cell protein was required. None of our unaffected control cell lines in the archives was suitable for this purpose so we needed to collect
fresh skin biopsies from healthy volunteers and establish new cultured fibroblast lines to be used as controls. Lipid rafts were then isolated from cultured skin fibroblasts from MPS patients and our healthy volunteers (controls) and the lipid composition was determined by mass spectrometry. In the raft domains we observed increases in the sphingolipid, ceramide and the glycosphingolipids, glucosylceramide, lactosylceramide and ceramide trihexoside, compared with our controls. Consequently, the lipid composition of the rafts was altered in MPS cells. In control cells, phosphatidylcholine was the major component comprising 45% of the lipid in the raft domain, whereas in the MPS cells, ceramide replaced phosphatidylcholine as the most abundant lipid comprising 35%, and reducing phosphatidylcholine to 20%. The Figure below shows the difference in the lipid composition between MPS I fibroblasts and controls.
Additional changes in the raft composition included increases in the glycosphingolipids with concomitant decreases in the phospholipids. Of note was an alteration in the anionic phospholipid bis(monoacylglycero)phosphate which we have previously shown to be elevated in MPS (Meikle et al., 2008). Therefore, from our studies in cultured fibroblasts we have shown that the lipid composition of rafts is altered in MPS. It is unclear at this stage what effects an altered composition of lipid rafts has on cell function and how it may lead to disease.
Figure: Lipid composition of rafts
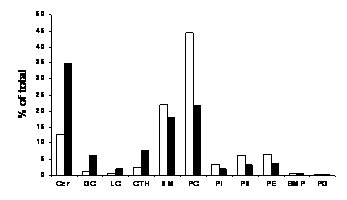
Lipid rafts were isolated from control skin fibroblasts (open bars) and MPS I fibroblasts (filled bars) and the lipid composition was analysed by mass spectrometry. The total amount of each lipid present in the rafts was expressed as a percentage of the total amount of all the lipids analysed.Cer, ceramide; GC, glucosylceramide; LC, lactosylceramide; CTH, ceramide trihexoside; SM, sphingomyelin; PC, phosphatidylcholine; PI, phosphatidylinositol; PS, phosphatidylserine; PE, phosphatidylethanolamine; BMP, bis(monoacylglycero)phosphate; PG, phosphatidylglycerol.
To mimic what effect enzyme replacement therapy may have on returning the lipid raft composition back to normal, the culture media of MPS I fibroblasts was supplemented with recombinant human α-L-iduronidase. Following 24 hr of treatment with the enzyme, the glycosphingolipids were starting to normalize. Further studies are underway with increased time and concentration of α-L-iduronidase to see if these glycosphingolipids can be completely normalized. Importantly, treatment with enzyme showed no reduction in ceramide or bis(monoacylglycero)phosphate. In unaffected cells we have shown that myriocin added to the culture media does reduce the amount of ceramide and further work is underway to test this in MPS cells. Additionally, lipid rafts are potentially modifiable by diet, particularly by dietary polyunsaturated fatty acids. Using control cells our initial results showed that supplementing the culture media with linoleic acid had no effect on the sphingolipids but did alter bis(monoacylglycero)phosphate. Further studies are underway to determine what effect linoleic acid has on MPS cultured skin fibroblasts, and whether combinations of drugs and fatty acids restore raft composition.
We are currently writing up our findings for publication and will send a copy of the manuscript to the society. We would like to thank the society for supporting our research and we hope to gain further independent funding to continue this work.
Meikle, PJ., Duplock, S., Blacklock, D., Whitfield, PD., Macintosh, G., Hopwood, JJ. and Fuller M. (2008) Effect of lysosomal storage on bis(monoacylglycero)phosphate. Biochem. J. 411, 71-78.
2006 Research Grant PDF Download
The National MPS Society has awarded $290,000 in new grants for 2006. One grant for MPS II and two grants for general MPS and Related Disease research were awarded. Each grant is for a total of $80,000, $40,000 for each of the two years of funding. An additional $50,000 was awarded to Dr. Brooks to further facilitate his work in MPS III with chaperone therapy.
Dr. Joseph Muenzer was awarded $120,000 in February, 2006 for his work “AAV Gene Therapy for Mucopolysaccharidosis II”. Dr. Muenzer will receive $60,000 for each of the two years of funding. Funding for this grant is from an MPS II grant not awarded in 2005 plus additional MPS II research dollars received in 2005.
Dr. Joseph Muenzer, University of North Carolina, Chapel Hill, NC
The focus of our laboratory is to develop new treatments for patients with MPS disorders. A mouse model for MPS II (Hunter syndrome) was created in the laboratory to develop gene therapy procedures to treat both the physical and the central nervous system involvement. The goal of gene therapy in MPS II is to replace the abnormal gene (iduronate sulfatase) by introducing a normal copy into cells. Modified viruses can be used to deliver genetic material into cells. We have created a delivery system using an adeno-associated virus (AAV) which contains a normal copy of the gene altered in MPS II. Our research has shown correction of the liver storage of glycosaminoglycans in MPS II mice after IV administration of these viruses.
This research proposal is to expand our studies in MPS II mice to characterize the extent of correction in other tissues and to use newer viruses that may allow better expression of iduronate sulfatase thereby improving the effectiveness of MPS II gene therapy. The successful completion of these studies will generate the animal data needed to submit an application for a human gene therapy clinical trial in MPS II patients.
Dr. Maria Pia Cosma, TIGEM, Naples, Italy
Children affected by mucopolysaccharidosis type II (MPS II: Hunter syndrome) lack the activity of the iduronate 2-sulfatase enzyme (IDS). They accumulate compounds in their body that gradually kill their cells and damage all of their visceral organs. At present, efficient cures are not available. A gene therapy approach was initiated to treat this disease in a mouse model of MPSII. The affected mice were injected with viral particles that targeted the liver. High levels of active IDS was produced and secreted into their plasma and taken up by all of the organs. This approach gave important results, as the mice were cured of their visceral defects. Now we plan to dissect carefully the brain defects in the MPSII mouse model. We have already generated data showing that we can deliver the IDS enzyme in the brain after systemic treatment of the MPSII mice. We now aim to design an efficient gene therapy approach to cure the brain defects in the mice thought direct deliver of the viral particles in the cerebrospinal fluid. Finally, our aim is to treat baby MPSII mice. Their liver and muscle will be modified to produce and secrete high amounts of active IDS into the blood, which can again cross-correct the other damaged organs. If successful, this approach should allow the treatment of the disease at a very early stage, and even before the symptoms are manifested.
The long term goal of this research is to establish a bone-targeting system for enzyme replacement therapy to improve the bone lesions present in the mucopolysaccharidoses (MPS). Current approaches for the treatment of MPS have little or no impact correcting the bone pathology. In this proposal, we will test a newly designed drug delivery system to target bone using an acidic amino acid oligopeptide as a carrier. Modifying the enzyme will allow it to be delivered to the bone more specifically and will enhance the drug effectiveness and reduce the side effects. The enzymes that are deficient in MPS will be tagged with the small acid peptide. The tagged enzymes will be tested on MPS mice models, initially MPS IVA, to evaluate whether it is more effectively delivered to the bone lesions compared to the untagged enzymes. By infusing the deficient enzymes into MPS IVA mouse models weekly for a long term, we can see how effectively they treat the disease and obtain helpful information required to design future human trials possibly applicable to all MPS diseases in which patients suffer from bone lesions. We are currently aiming at the human clinical trial on MPS IVA based upon the result.
Genetic diseases are a fundamental cause of human suffering, depriving many children (up to 2.5% of births) and some adults of a normal existence. Lysosomal storage disorders (LSD) are a group of over 45 different genetic disorders, each involving a defect in lysosomal function. Mucopolysaccharidosis III A (MPS III A) is an LSD that affects mainly children, but also some adults and can result in very severe brain disease. MPS II A is caused by a defect in an enzyme called sulphamidase. We plan to develop a new therapy, which will involve stabilising the patient’s mutant sulphamidase protein, to increase the level of enzyme activity and treat the disease. We will test various chemical compounds for their ability to increase the level of sulphamidase enzyme activity in patient cells. The project will potentially lead to the development of a safe, non-invasive therapy to treat this severe disorder.
“AAV-mediated gene therapy of Hunter syndrome in the MPS II mouse model”
TIGEM, Naples, Italy
Mucopolysaccharidosis type II (MPSII; Hunter syndrome) is a lysosomal storage disorder that arises due to the deficiency of iduronate 2-sulfatase (IDS) enzyme activity. We recently characterized the ids knockout, which shows skeleton deformations and an elevated accumulation of glycosaminoglycans in the urine and in many of organs. In addition, the performance of the knockout mice in the open field and walking pattern tests were severely compromised. We designed an efficient gene therapy approach to treat these MPSII mice using adeno-associated viral (AAV) vectors. AAV2/8TBG-IDS viral particles were administrated intravenously in adult animals. The plasma and tissue IDS activities were completely restored in all of the treated mice, up to nine months after treatment. This rescue of IDS activity resulted in the full clearance of glycosaminoglycan accumulation in the urine and in all of the tissues analyzed, with correction of the skeleton malformations and normalization of the performance in the locomotor tests (Cardone et al. Hum Mol Genet 2006). It is gratefully acknowledged that these studies were made possible through funding provided by the National MPS Society Award 2004. With regard to the current MPS Award 2006, to date, our progress towards the aims proposed is as follows:
Aim 1: Characterization of the brain defects in MPSII mice.
We have performed an initial characterization of the brain defects in the MPSII mouse model and seen a loss of Purkinje cells and cellular vacuolization in different regions of the brain: the hippocampus, thalamus, cerebellum and brainstem. We also noted GAG accumulation within the choroids plexus of the ventricular region. To further characterize the brain abnormalities in MPSII mice, we planned to analyze the morphology and numbers of neurons, astrocytes and oligodendrocyte, using different neural markers. To begin this analysis, control wild-type adult mice and MPSII adult mice, all at 12 months of age, were perfused with PFA and the brains postfixed and embedded in paraffin. Sections were analyzed by immunohistochemical analysis using NeuN as the neuronal cell marker, calbindin as the Purkinje cell marker and GFAP as the astrocyte marker. We detected a decreased number of NeuN-positive neurons in the cortex and almost no positive calbindin signal in the cerebellum of the MPSII mice, as compared to the wild-type animals. In contrast, we detected an increased positive GFAP signal in the astrocytes of the MPSII brain sections. These preliminary data suggest that neurodegeneration and gliosis occur in the brains of adult MPSII mice.
Aim 2: Development of a gene therapy approach for the treatment of the MPSII pups to anticipate disease manifestation.
Preliminary short-term experiments have indicated that when the idsy/- pups are injected with the AAV2/5CMV-IDS and AAV2/8TBG-IDS vectors into the temporal vein, they show a rescue of IDS activity and a clearance of GAG accumulation in all of the organs analyzed, including the brain. To see whether it is possible to prevent the MPSII phenotype, newborn (2 days after birth) idsy/- pups received 1×1011 total particles of AAV2/5CMV-IDS, of AAV2/8TBG-IDS or of AAV2/5CMV-EGFP in the temporal vein. We plan to follow the efficacy of the therapy throughout their lifetime. We are measuring IDS activity in the plasma and GAG accumulation in the urine each month. The IDS-injected mice are now 13 months old and show very high levels of circulating enzyme in the plasma, and GAG clearance in the urine, with respect to the control group.
In addition, we planned to test the efficacy of the intra-muscular therapy. For this, we have constructed the AAV2/1MCK-IDS vector. The AAV2/1 serotype in combination with the muscle creatinine kinase promoter (MCK) allows efficient muscle transduction and robust expression of the transgene. We tested the prepared vector in wild type mice. Thus, a group of wild-type animals have been injected in the anterior tibialis with 2×1010 total particles of AAV2/1MCK- IDS. The injected animals were sacrificed one month after the injection and the transduced muscles were harvested. The measured IDS activity was extremely high in the injected muscle, with respect to the activities measured in the control (non-injected) group.
Aim 3: Development of a gene-therapy approach to correct the CNS defects of the MPSII mice. To treat the neuropathological features via systemic delivery of the viral particles, we constructed the AAV2/4CMV-IDS vector and disseminated it into the cerebro-spinal fluid through injection into ventricular region IV. The goal of this approach is to transduce CNS ependymal cells and test if these cells, once transduced, can serve as a source for enzyme secretion into the surrounding brain parenchyma and CSF. A group of with 1×1010 particles of AAV2/4CMV-IDS or with 1×1010 of AAV2/5CMV-GFP viral particles as control. The mice were sacrificed one month after treatment and the brains analyzed. An increased IDS activity was seen in the brain homogenates of IDS-injected mice, with respect to the controls. In addition clearance of GAG accumulation was the brain sections of the treated mice.
“Development of a therapeutic bone-targeting system for MPS”
Department of Pediatrics, Saint Louis University, Pediatric Research Institute
We have finished the untagged enzyme experiment on adult and newborn MPS IVA mice. The tagged enzyme is now under investigation. We summarized the results as follows. In the second year, we will finish the bone-tagged enzymes as well.
Summary of the results:
The half-lives in blood circulation of two phosphorylated GALNS enzymes were similar (native,2.4 min; SUMF1, 3.3 min). After intravenous doses of 250 units/g body weight were administered, each enzyme was primarily recovered in liver and spleen, with detectable activity in other tissues including bone and bone marrow but not in the brain. At 4 h postinjection, enzyme activity was retained in the liver, spleen, bone, and bone marrow at levels that were 20% – 850% of enzyme activity in the wild-type mice. After intravenous doses of 250 units/g of native GALNS, 250, 600, or 1,000 units/g of SUMF1-GALNS were administered weekly for 12 weeks, MPS IVA mice showed marked reduction of storage in visceral organs, bone marrow cells, osteoblasts, osteocytes, ligaments, and periosteum. A dose-dependent clearance of storage material was observed including brain and cartilage cells although the heart valves were refractory and variable. The blood KS level assayed by tandem mass spectrometry was reduced nearly to normal level. These preclinical studies demonstrate the storage clearance of tissue and blood KS by administered GALNS, thereby providing the in vivo rationale for the design of ERT trials in MPS IVA. Preliminary results by using the bone-targeting enzymes showed that intravenous doses of 250 units/g of the bone targeting GALNS was the same effective as 1,000 units/g of SUMF1-GALNS was used.
We have used knock-out MPS IVA mice to assess the effects of long-term enzyme-replacement therapy initiated at birth. MPS IVA mice received weekly i.v. injections of 250 units/g body weight recombinant human native or SUMF1-GALNS until 14 wk of age. Either GALNS is able to reach brain and bone until the blood-brain barrier completely closes at 10�”14 d of age and avascular region in cartilage cell layer appears. MPS IVA mice that were treated from birth demonstrated near normalization or complete reversal of lysosomal storage in most tissues including bone marrow, bone (osteocytes, osteoblasts, periosteum, and cartilage), ligaments, and heart valves. Nearly absence in storage vacuoles in cells of the CNS in MPS IVA mice treated from birth was also observed. MPS IVA mice treated from birth kept normal level of serum KS significantly lower than untreated MPS IVA mice cells. Preliminary results by using the bone- targeting enzymes showed that intravenous doses of 250 units/g of the bone targeting GALNS was the same effective as native or SUMF1-GALNS was used.
These data suggest that enzyme that enters the brain and the cartilage in the first few weeks of life, before the blood-brain barrier and cartilage cell layers mature, is able to protect against accumulation of storage material in MPS IVA mice.
“Evaluation of Enzyme Enhancement Therapy for Mucopolysaccharidosis Type IIIA”
Women�™s and Children�™s Hospital North Adelaide, South Australia Australia
Genetic diseases are a fundamental cause of human suffering, depriving many children (up to 2.5% of births) and some adults of a normal existence. Lysosomal storage disorders are a group of over 45 different genetic disorders, each involving a defect in lysosomal function.
Mucopolysaccharidosis IIIA is a lysosomal storage disorder that affects mainly children, but also some adults and can result in very severe brain disease. Mucopolysaccharidosis IIIA is caused by a defect in an enzyme called sulphamidase. We plan to develop a new therapy, which will involve stabilising the patient�™s mutant sulphamidase protein, to increase the level of enzyme activity and thus treat the disease. To increase the stability of the mutant sulphamidase protein we will evaluate small molecule compounds called chemical chaperones that either interact with the outside of the protein or help stabilise the protein from within, by interaction with either the core of the molecule or the active site of the protein.
In October 2006 we commenced this project by appointing an experienced research officer for this research project. Because of the drive to develop a new therapeutic strategy for mucopolysaccharidosis IIIA, we first set about validating all of the assay systems that would be required to convince a regulatory authority of the soundness of any data generated. We also cultured a number of cell lines expressing point mutations in CHO-K1 cells and selected fibroblast cell lines that would be appropriate for evaluating the action of chemical chaperones. These cell lines will be used to show increased protein stability and amount of protein/activity in response to chemical chaperone treatment. These initial objectives were completed early in 2007 and showed that the sulphamidase assays were both accurate and not subject to significant variation.
We have now commenced a preliminary screen of the compounds proposed as chemical chaperones. In previous work, we showed that protein stabilisers like glycerol and trehalose (that act on the outside of a protein) can improve the yield of mutant protein and activity, but for sulphamidase we have seen no affect in expression cell lines that had different point mutations. We then evaluated several compounds that would be expected to bind to the inner core of the molecule, to help stabilise the mutant sulphamidase protein. We evaluated glucosamine (a low affinity inhibitor of sulphamidase) and glucosamine N-sulphate and showed some variable increases in the sulphamidase protein. This may indicate a partial effect, or that the conditions that we used were not optimal and we are currently pursuing this line of investigation. Finally, we have just evaluated another different type of inhibitor and have some indications of a very promising result. Thus, we believe that we are on target to achieve the following two scientific aims later in 2007:
Identification of a chemical chaperone that will enhance the level of mutant sulphamidase protein and enzyme activity in cultured cells.
Demonstrate that chemical chaperone treatment can correct the MPS IIIA biochemical storage defect, in vitro.
“AAV-Mediated Gene Therapy of the Hunter Syndrome in the MPS II Mouse Model�?
Received 7-08
Mucopolysaccharidosis type II (MPSII; Hunter syndrome) is a lysosomal storage disorder that is due to a deficiency of the iduronate 2-sulfatase (IDS) enzyme activity. We have characterized the ids knock-out, which shows skeleton deformations and an elevated accumulation of glycosaminoglycans (GAGs) in the urine and in many organs.
More recently, we have started the characterization of the brain phenotype of the knock-out mice. Control wild-type mice and three-month-old adult MPSII mice were analyzed by immunohistochemistry using NeuN as a neuronal marker, calbindin as a Purkinje cell marker, and GFAP as an astrocyte marker. Furthermore, we also tested ubiquitin abundance as a marker for neurodegeneration. In the MPSII mice, we detected a decreased number of NeuN positive neurons in the cortex and a strong reduction in the Purkinje cells in the cerebellum, according to the reduction in the calbindin signal. In contrast, we detected an increase in the GFAP signal and an increase in the ubiquitin-positive signal in the neurons of the MPSII brain sections, with respect to the wild-type mice. These data suggest that neurodegeneration and astrogliosis occurs in the brain of the adult MPSII mice.
We have also performed neurobehavioural tests to analyze the impairments of selected behavioural domains. Groups of control wild-type and MPSII mice of 4.5 and 5.5 months were analyzed. The results have revealed that MPSII mice have an abnormal fear response in the elevated plus maze, spending longer times on the open arms than the control mice, and a memory deficit in the novel-object recognition test, exploring novel and familiar objects equally. These findings show that the emotional and cognitive behavioural domains are impaired in the mutant mice, which are likely to be the behavioral domains that are homologous to those affected inHunter patients.
Regarding therapeutic strategies, we have already designed an efficient gene therapy approach to treat these MPSII mice using adeno-associated viral (AAV) vectors. AAV2/8TBG-IDS viral particles were administered intravenously in the adult MPSII mice. The plasma and tissue IDS activities were completely restored in all of the treated mice up to thirteen months after treatment. This rescue of IDS activity resulted in full clearance of GAGs accumulation in the urine and in all of the tissues analyzed, with correction of the skeleton malformations and normalization of the performance in the locomotor tests (Cardone et al. Hum Mol Genet 2006).
We have now performed systemic delivery of AAV2/5CMV-IDS vectors, with the aim of treating both visceral and CNS defects and to anticipate disease manifestation. We have carried out experiments where we have treated MPSII pups at the newborn stage. We injected groups of MPSII pups in the temporal vein with 1×1011 total particles of AAV2/5CMV-IDS. The mice were sacrificed 1 month and 1.5 years after this injection. The activities of IDS in all of the tissues reached levels that were higher then those in the group of non-treated MPSII mice.
Remarkably, IDS activity was also increased in the brains of the treated mice. The GAG accumulation analyzed in sections was fully cleared in the visceral organs and also in all of the brain areas analyzed (choroid plexus, meninges, cerebellum, cortex) in both groups of treated mice (1 month and 1.5 years after injection). Finally, immunohistochemistry using an anti-GFAP antibody showed a clear amelioration of the astrogliosis phenotype in both groups of treated mice. These data demonstrate that systemic administration of viral vectors carrying the IDS enzyme can ameliorate both the CNS and the visceral symptoms of MPSII mice.
In addition, we are developing a direct CNS-therapy approach. A group of MPSII mice was injected at three months of age with 1×1010 particles of AAV2/4CMV-IDS, or with 1×1010 of AAV2/5CMV-GFP viral particles as the control. The goal of this approach is to transduce CNS ependymal cells and determine if once transduced, these cells can serve as a source for enzyme secretion into the surrounding brain parenchyma and the CSF. The mice were sacrificed 1 month after treatment and the brains were analyzed. Increased IDS activity was seen in the brain homogenates of these IDS-injected mice, with respect to the controls.
University of North Carolina School of Medicine, Chapel Hill, NC
“AAV Gene Therapy for Mucopolysaccharidosis II�? review not received
Department of Pediatrics, Saint Louis University, St. Louis. MO
“Development of a Therapeutic Bone-Targeting System for MPS�? review not received
Women’s and Children’s Hospital, North Adelaide, South Australia, Australia “Evaluation of Enzyme Enhancement Therapy for Mucopolysaccharidosis Type IIIA�? received 11-08
Mucopolysaccharidosis IIIA (MPS IIIA) is a lysosomal storage disorder that primarily affects the central nervous system, leading to progressive loss in brain function. MPS IIIA is caused by a defect in the activity of an enzyme called sulphamidase. Most patients with MPS IIIA have sulphamidase present, but it is either in an amount that is insufficient to enable it to carry out its normal function, or it is in a form that is not active. With the generous support of The National MPS Society this project sought to evaluate a new therapy for MPS IIIA, which centres on the concept of using chemicals to stimulate [or ‘enhance’] the sulphamidase that is present in patients and guide [or ‘chaperone’] it through a series of cellular processes to enable therapeutic benefit. We have investigated a range of compounds or ‘chemical chaperones’ that can potentially interact with sulphamidase to increase its stability and function.
The main aim of this two year project was to test a number of possible chemicals for their capacity to enhance the activity of sulphamidase present in cultured cells, enabling us to identify an optimal compound for further testing in an animal model of MPS IIIA.
In the first year we screened a number of potential chemical chaperones in human MPS IIIA cultured hamster ovary cells that expressed different mutations [the inherited mistakes] that cause MPS IIIA. Compounds such as glycerol and trehalose that tend to act as general stabilisers
showed little or no effect on enhancing the activity of the sulphamidase present in these cells. We also tested the compounds ortho- and meta-vanadate, which resemble a specific stage in normal substrate [the material stored in MPS IIIA patients] breakdown but these also showed little or no effect. Next, we evaluated a panel of six compounds that can bind to the active site of sulphamidase. Several of these, including glucosamine, had no effect, but two were found to have an enhancement effect on sulphamidase in cultured cells.
In the second year these two candidate compounds were evaluated in MPS IIIA mouse and human cultured skin cells and showed similar results. Unfortunately, the project was interrupted for six-months at this stage due to the departure of the scientist working on the project. In recent experiments, we observed a dramatic enhancement in sulphamidase activity in cells treated with one of the two candidate compounds, but this effect was not reproducible, suggesting that either the chemical dose or experimental conditions were not optimal. We are continuing to optimise the dosage/conditions for this test compound to enable the evaluation of this or a related compound in animal model experiments.
We thank the Society for its support of this project.
2005 Research Grants PDF Download
The National MPS Society has awarded $320,000 in new grants for 2005-2006. Two grants for MPS III research were awarded $60,000 each, $30,000 for each of the 2 years of funding. This year a special grant category was created for research specific to Bone and Joint research, and those researchers will receive $50,000 for each of the 2 years of funding, for a total per grant of $100,000. The MPS II research grant offered this year will not be funded due to the lack of acceptable proposals received.
The blood brain barrier is the single greatest hurdle to bringing therapies for neuropathic lysosomal storage diseases to clinical application. One approach to overcome this barrier involves direct injection into the central nervous system of either recombinant enzyme, gene therapy vectors, stem cells, or gene therapy treated stem cells. An alternate method is to genetically engineer the missing enzyme of a particular lysosomal storage disease, so that it is able to cross the blood brain barrier by an active process of uptake from the blood and delivery across the blood brain barrier. This would allow for treatment of the central nervous system either by intravenous delivery of the engineered recombinant enzyme, or alternatively, treatment with a liver directed gene therapy vector designed to deliver the gene for the engineered enzyme. We propose to evaluate this latter method to develop a treatment for mucopolysaccharidosis type IIIB (MPS IIIB). Using parts of proteins that bind the LDL receptor we propose to genetically engineer the Naglu enzyme so that it will cross the blood brain barrier via this receptor system. We will evaluate this method n a cell culture system, and successfully engineered enzyme will then be evaluated clinically using liver directed gene transfer into the murine and canine models of MPS IIIB.
Australia
“Lentiviral-mediated Gene therapy for MPS IIIA”
Gene therapy is an attractive option for the treatment of the mucopolysaccharidosis (MPS) including MPS III. The development of a new generation of technologies for the delivery of genes has meant that gene therapy now looks a more realistic option than ever before. However, the safety and efficacy of these technologies need to be carefully evaluated before clinical application can be considered, and even with these new vectors, treatment of brain pathology is challenging. This project aims to use a small animal (mouse) model of one of the most common MPS, MPS III A, to evaluate a new gene delivery technology for its ability to treat brain pathology. Two methods for delivering the vector to the bran will be evaluated. The level and distribution of gene delivery that results, its persistence, and its effect on disease pathology, will be assessed using a variety of enzymatic, molecular and biological techniques.
The overall goal of our work is to use animal models of the mucopolysaccharidoses (MPS) to investigate the underlying causes(s) of cartilage and bone disease in these disorders. We have already shown that the cells in MPS cartilage are prone to death, and that in response to this primary damage, a series of biochemical changes occur (known as ?inflammation?), exacerbating the disease. We will continue to evaluate the causes(s) of MPS bone and joint disease, and based on these results, identify new ways to monitor disease severity, progression and treatment response. We will also evaluate new approaches to therapy. We will extend our findings in the cartilage of MPS animals to other joint tissues and bone. We will assess cell death and ?inflammation? in these tissues, as well as study bone growth. We will also utilize findings from these studies to identify specific ?biomarkers? that might predict disease severity and treatment response. For this we will study these markers in the blood and joints of MPS VI animals treated by enzyme replacement and gene therapy. Lastly, we will use MPS animal models to evaluate new approaches to treat these disorders, including using inhibitors of?inflammation? and/or cell death, as well as enzyme replacement therapy using a new form of the recombinant enzyme that can penetrate cartilage more efficiently.
Australia
“The Pathogenesis of Cartilage Degradation in MPS VI”
Cartilage/joint disease presents a significant challenge to the clinical care of patients with mucopolysaccharidosis (MPS). With the advent of enzyme replacement therapy (ERT), the injection of enzyme into the circulation and into the localized joint space has the potential to significantly alter the progression of cartilage disease in MPS. However, to maximize the benefit of ERT it must be applied prior to the start of clinical symptoms. The benefit of ERT to patients with established symptoms is less clear cut. The pathogenesis of cartilage destruction in MPS I is poorly understood although it appears to resemble that observed in osteoarthritis (OA). In particular the turnover of the glycosaminoglycan (GAG) component of MPS cartilage and the relationship between GAG storage and disease progression is unclear. The focus of this proposal is to understand the mechanism of cartilage destruction in MPS, using the MPS VI cat model.
The degradation of cartilage proteoglycans and the subsequent uptake and storage of their GAG component will be investigated. This information will be used to develop supplemental therapies targeted to joint disease in MPS.
1st Year Research Reviews
The immediate aims of this proposal are to (i) characterise the breakdown products of cartilage proteoglycan in MPS VI with specific reference to the fragments generated by aggrecanase or matrix metalloproteinase activity (MMP) and to (ii) assess proteoglycan turnover in MPS VI cartilage cells. The long-term goal of the proposal is to determine if cartilage degradation in MPS VI follows a pathway similar to that observed in osteoarthritis (OA) or rheumatoid arthritis (RA). If this correlation can be made it will open the way to apply OA or RA therapies to MPS VI as a supplement to systemic treatments such as ERT in an effort to specifically target joint disease.
We have made significant progress towards aim (i) with the identification of an MMP driven cartilage turnover process. Normal and MPS VI cartilage from 2, 6 and 11 month old cats was extracted into 4M guanidine HCl buffer and digested with chondroitinase ABC prior to electrophoresis on SDS-PAGE. Western blot analysis was carried out using a panel of antibodies directed towards the carbohydrate or protein components of intact proteoglycan or towards neo-epitopes generated by aggrecanase or MMP cleavage of proteoglycan. Total proteoglycan analysis demonstrated predominantly aggrecan with lesser amounts of decorin/biglycan that did not significantly alter between normal and MPS VI or with age. The presence of low levels of an ITEGE neo-epitope indicative of aggrecanase digestion of aggrecan was observed in both normal and MPS VI cartilage that did not alter with age. In contrast, a DIPEN neo-epitope indicative of an MMP directed digestion of aggrecan accumulated with age. Gelatin zymography demonstrated the presence of detectable levels of MMP-9 in cartilage from 2 month old MPS VI cats. MMP-9 did not become apparent in normal cartilage until 6 months of age or later. MMP-2 levels were also increased in MPS VI although not to the same extent.
MMP-3 and MMP-7 were not detected in either normal or MPS VI cat cartilage.
In degenerative joint diseases such as OA or RA, MMPs degrade both the proteoglycan and collagen component of cartilage leading to irreversible tissue damage. Our initial results suggest that the same mechanism may be involved in MPS VI joint disease and that MMPs would constitute a potential intervention point.
Staff: Ms Lann Tay, summer student and Ms Chantelle McIntyre, PhD student
Lentiviral vectors represent highly modified viruses that can be used to introduce gene sequences into cells. In this project a lentiviral vector will be used to introduce a normal copy of the sulphamidase gene, which is defective in MPS III-A, into the liver and the central nervous system of MPS III-A mice. The effect of gene transfer on the course of the disease will then be monitored. This will include assessment of brain function as well as histopathology and biochemical analyses of tissue samples.
Ms Lann Tay, a third year student from the University of South Australia, was funded from the grant. Ms Tay worked on finalising a lentiviral vector construct that is to be used in the study. We hope to attract Ms Tay back as an Hons student in 2007.
In order to maximise the return from the funding received from the MPS Society, and to have a full time person work on the project, we have established a postgraduate scholarship award in conjunction with the University of Adelaide. Ms Chantelle McIntyre was awarded this scholarship and started work on the project in January of this year. Ms McIntyre is a very able and focussed student and she has quickly established the project and the work is now moving rapidly forward.
Ms McIntyres work has initially focussed on finalising initial studies on somatic lentiviral- mediated gene therapy in the MPS IIIA mouse. This work had two clear aims
The results of (2) have shown that the elongation factor 1a gene promoter is significantly better (approximately 3-fold higher expression) than the pgk promoter originally used. Accordingly this promoter will be used for all further studies. As it has been widely established that the efficacy of gene therapy for the storage diseases (by whatever route) correlates with the level of enzyme expression we are also exploring other means of enhancing sulphamidase expression from our vectors including codon optimisation of the sulphamidase coding sequence and the use of RNA transport elements in the sulphamidase transcript.
The further analysis of samples from the MPS IIIA mice treated with the LV-pgk-msulp construct has confirmed the efficacy of intravenously delivered vector for treating much of the somatic disease. More interestingly, our results suggest that this approach may also be efficacious for treating central nervous system pathology. We have found that the elevated level of b-hexosaminidase expression seen in MPS IIIA central nervous system tissue is significantly reduced in the treated mice. b-hexosaminidase levels are widely accepted as a measure of pathology in the storage disorders. Unfortunately, sulphamidase levels could not be measured directly in central nervous system tissue from treated mice due to the limited sensitivity of the sulphamidase enzyme assay. However, our result is not without precedent, AAV-mediated liver targeted gene therapy (1) and high dose enzyme replacement therapy studies (2) in the MPS VII mouse have both indicated that enzyme transport across the blood brain barrier does occur in adult mice, albeit at low levels. These results suggest that high level expression of sulphamidase in somatic tissues such as the liver has the potential to be efficacious in treating the central nervous system as well as poorly vascularised somatic tissues/cells such as chrondrocytes. We are currently trying to confirm these results by direct measurement of storage material in the central nervous system using mass spectrometry. We feel this is an exciting result as it raises the possibility of a relatively non-invasive gene therapy approach for MPS IIIA central nervous system disease. This study is currently being prepared for publication and will be presented at the 2006 International MPS Meeting in Venice by Dr Sharon Byers.
We have also established the stereotaxic injection equipment necessary for the direct injection of virus into the CNS. We are currently preparing virus for our first experiments and anticipate that these will take place in the next 4-6 weeks.
In addition, we have imported the MPS IIIA strain from Jackson labs (Sgshmps3a/PstJ) in order to establish a colony that has been backcrossed onto on a pure genetic background (C57BL/6J) that is more suitable for our research purposes (removing genetic variation) than our previous colony (the original “New York” strain) that was on a mixed (129SvJ, C57BL/6, SJL, CD1) genetic background. These mice have now cleared quarantine.
The CIB, Dr Sharon Byers, has now established that a modified water maze, the T maze, offers the most robust way of assessing neurological function, accordingly, we will be using this test in all our future studies in the MPS IIIA mice.
Our priorities for the second year of finding are to
The object of this research proposal is to generate a fusion of the enzyme Naglu, that will be able to cross the blood brain barrier by active transcytosis, leading to a CNS treatment of MPS IIIB. Such an enzyme fusion could be used for either gene therapy, or intravenous enzyme replacement therapy. The strategy we propose is to creat fusions between Naglu and and the ligand domains of the LDL receptor ligands Apolipoprotein B and E (ApoB and ApoE). The grant has 3 specific aims.
Aim 1 is to construct and evaluate the enzyme fusions in vitro.
Aim 2 is to evaluate the behavior of the fusions in vivo using mice and an AAV2/8 vector. Aim 3 is to evaluate efficacious vectors from Aim two in the canine model.
To date we have constructed three Naglu enzyme fusions, one with the ApoE at the beginning of the enzyme (amino terminus fusion), and one each with ApoB and ApoE at the end of the enzyme (carboxy terminus fusions). Our results indicate that, compared to the normal human Naglu enzyme, the amino-terminus ApoB-Naglu fusion protein expressed in human embryonic kidney cells (293 cells) produce enzymatic activity of 60% whereas the carboxy-terminus Naglu-ApoB and Naglu-ApoE fusions produce about 30% of activity. Moreover, the Naglu activity was also detected in the media of the transfected cells suggesting that the ApoB and ApoE fused Naglu proteins are secreted in the cell culture media. The expression of the recombinant ApoB and ApoE Naglu fusions was confirmed by western blot using an anti- human Naglu specific antibody and an antibody which recognized a tag sequence of the fused domain. We also performed immunofluorescence cell staining to confirm that our recombinant constructs yield intact enzyme fusions able to localize in the lysosomes. Immunofluoresent cell staining with confocal microscopy demonstrates that the myc signal (a tag element in the fusions) in transfected primary normal dog fibroblast co-localize with both an anti-human Naglu antibody and a lysosome specific anti-Lamp1 antibody. These results suggest that significant Naglu activity is still present if fused with the ApoB ligand domain at its amino-terminal extremity and that the Naglu activity is higher with an amino-terminus fusion than with a carboxy fusion.
Further work underway at this time includes the purification of enzyme fusions to measure the specific activity or the enzyme fusions relative to normal human Naglu. Additionally, AAV2/8 vector constructs are being designed to express these fusions and will be evaluated using a liver expression/gene therapy approach in the MPS IIIB knockout mouse. These studies will form the preliminary study for a gene therapy evaluation of this approach using the MPS IIIB dog.
Work supported by this grant was presented this April (2006) in Alexandria, Virginia at the NINDS Workshop: Glycosphingolipids in Health & Disease, and at the 9th International Symposium of Mucopolysaccharide and Related Disorders, in Venice Italy, June, 2006.
The entire amount of this award ($30,000) has gone toward the salary and benefits support of Dr. Rafi Awedikian (~80% supported on MPS Society funds).
The major goal of our research is to gain a better understanding of bone and joint disease in the MPS disorders, and, based on these findings, to develop improved therapies for these organs.
We are also attempting to identify improved biomarkers that can be used to predict the severity of bone and joint disease in individual MPS patients, as well as their response to therapy.
During the past year we have demonstrated an important connection between the immune and skeletal systems in the MPS disorders. In addition to our previous results showing that cells in the joints and bones of MPS animals undergo premature cell death due to the accumulation of glycosaminoglycans (GAGs), our new studies suggest that an inflammatory disease is occurring in these animals similar to rheumatoid arthritis.
In MPS and arthritis, the stimulation of inflammatory cytokines (IL-1b and TNF-a), proteins that stimulate or inhibit the proliferation or function of immune cells, leads to the secretion of several matrix metalloproteinases (MMPs), which in turn lead to cartilage degradation and, ultimately, to bone destruction. The activation of cells of the synovial membrane, which lines the cavities of joints, as well as specialized immune cells, or T cells, have a positive effect on osteoclast (bone absorbing cells) formation by stimulating certain blood cells (macrophages) to secrete additional proinflammatory cytokines into the joint space. The production of these cytokines leads to the differentiation and activation of osteoclasts, resulting in bone loss and joint destruction. These findings were not restricted to one particular MPS disorder or species, and appear to define a common, broad mechanism of bone and joint destruction in these disorders. The use of anti-inflammatory agents to block the secretion of proinflammatory cytokines at various stages in the inflammatory cascade is presently being investigated in our lab. This therapeutic strategy might be used as a combination therapy with ERT to alleviate the bone and joint disease in MPS.
In general, our preliminary results indicate that early in life, an abnormal cellular and molecular profile is seen in MPS bones and joints, with characteristic increases in cytokines, metalloproteinases (MMPs), and dead (apoptotic) cells, principally in joint and bone growth plate cells (chondrocytes), synovial membrane, and synovial fluid. There is an important stimulatory (chemotactic and pro-inflammatory) role for macrophage inflammatory proteins (MIPs) in the development of these lesions, and the formation of multinucleated osteoclast-like cells (MNCs). The production of the cytokine, TNF-a up-regulates an essential osteoclast differentiation/survival factor, the ligand of receptor activator NF-kB (RANKL), and is a likely explanation for the appearance of the MNCs, which may result in clinical osteopenia (bone loss) in MPS. RANKL is produced by bone marrow stromal cells, osteoblasts, and synovial fibroblasts.
Normally, RANKL resides in cell membranes, and exerts its effect by direct interaction of bone cells (e.g., stromal cells/osteoblasts with osteoclast precursors), which express its receptor, RANK. RANKL is an essential factor for full differentiation and activation of osteoclasts. Thus, a consequence of having too much RANKL would be over-proliferation of osteoclasts, leading to bone destruction. Many of the cytokines known to stimulate bone resorption, such as IL-1b and TNF-a, act through up-regulation of RANKL. In arthritis, RANKL mobilizes osteoclasts causing bone degradation. In the context of inflammation, TNF-a induces RANKL synthesis by marrow stromal cells, and prompts TNF-a expression by osteoclast precursors. Thus, RANKL is not only involved in osteoclast proliferation, but also pathological bone loss. In MPS bone marrow cultures, our preliminary results indicate there is an increase in osteoclasts. These results, combined with our previous findings, suggest that cooperation of TNF-a and RANKL plays an important role in inflammation and bone destruction. Therefore, it is important to evaluate treatment strategies aimed at mediating the effects of TNF-a.
MIP-1a(macrophage inflammatory protein) is another pro-inflammatory molecule that binds to the cell surface CC chemokine (chemoattractant for white blood cells, or monocytes) receptors. MIP-1 proteins recruit pro-inflammatory cells (neutrophils, eosinophils, monocytes) to the site of injury, and are crucial for T-cell recruitment from the circulation to inflamed tissue.
Therefore, MIP-1 proteins are key players in the pathogenesis of many inflammatory conditions, including asthma and arthritis. It has also been shown that this chemokine acts as a recruitment factor for mature osteoclasts, as well as osteoclast precursors. RANKL-dependent proliferation of osteoclasts is up-regulated by MIP-1a In MPS animals and patients, up- regulation of these osteoclasts could occur by RANKL and MIP-1a secretion in the area of bone destruction. We have detected MIP-1a positive cells in the marrow cavity of MPS animals.
We have also evaluated the proteases, MMP-2, -9, and 13, in articular cartilage, growth plate cartilage, synovial fluid, and synovial membrane, gelatinase activity in synovial membrane, tartrate-resistant acid phosphatase activity for osteoclasts in bone marrow, and IL-1b in growth plates from normal and MPS animals. All of these compounds are involved in bone and joint homeostatis and reactions to injury. The pattern of changes from normal in these parameters in MPS animals will provide insight into the pathogenesis of the bone and joint lesions, identify targets for therapy, and leads to biomarkers that can be used to determine the effects of treatment.
Summary: Overall, we have found that GAG accumulation in MPS animals stimulates a number of pathological processes. These include enhanced death (apoptosis) of bone and joint cells, as well as inflammation. The inflammatory response, in turn, leads to proliferation of bone absorbing cells (osteoclasts), which leads to bone destruction. In general, many of the pathologic processes in the MPS bones and joints have close similarities to those that occur in arthritis. Thus, some of the important biomarkers and therapeutic targets for arthritis should be considered for MPS. Indeed, we have recently found that several of these markers are abnormally expressed in MPS animals. These include TNF-a, MIP-1, MMPs, RANKL, and others.
2004 Research Grant PDF Download
Eight human genetic diseases are caused by a deficiency in the enzymatic activity of sulfatases. Among these are five different types of mucopolysaccharidoses (MPS types II, IIIA, IIID, IVA, and VI). The final goal of this project is to develop a gene therapy approach to treat the MPS II (Hunter syndrome) which is due to the deficit of the Iduronate sulfatase (IDS). To this end, we will use a mouse model of MPS II which exhibits many of the characteristics of the human disease. IDS, as well as all the other sulfatases, need to be modified in the cell after they are synthesized in order to be active. We have recently identified SUMF1 gene (Sulfatase Modifying Factor 1), which is responsible for this modification. SUMF1 has a striking enhancing effect on the activity of all sulfatases in cultured cells. To increase our chances of having active IDS in vivo we will deliver both IDS and SUMF1 genes in the MPS II mouse model. The simultaneous delivery of SUMF1 and IDS genes should result in a more effective treatment of MPS II. Ultimately, this protocol could set the stepping stone for the treatment of other MPS due to sulfatase deficiencies.
Characterization of the Systemic Inflammatory Response to Lysosomal Storage? Dr. Mark Sands, Washington University School of Medicine, St. Louis, MO
Our laboratory is studying several mouse models of lysosomal storage disease that have characteristics similar to the human disease. While we were studying the MPS I mouse, we discovered that the mice have anemia and have difficulty putting on body weight as fat. Our curiosity about this lack of fat led to a variety of new studies. The MPS I mice eat the same and absorb the same amount of nutrients as unaffected mice. MPS I mice also have the same respiration and activity levels as normal mice, thus they have similar metabolic rate. Both the anemia and fat loss could be caused by abnormal inflammatory or immune responses. We therefore looked for inflammation markers in the blood and found an increase in the amount of inflammatory proteins in the affected mice. We are currently determining if the anemia, fat loss, and inflammation are common to other lysosomal storage diseases. Our ultimate goal is to determine what role the inflammatory proteins are playing in lysosomal storage diseases.
Research Updates
In 2004 the National MPS Society awarded grants to Dr Maia Pia Cosma for her work, ?Gene Therapy for MPS II? and to Dr. Mark Sands for his work ?Characterization of the Systemic Inflammatory Response to Lysosomal Storage?. Below are the reviews they submitted, summarizing the first year of research.
?Gene Therapy for MPS II?
Mucopolysaccharidosis type II (Hunter syndrome) is due to the deficiency of the enzyme iduronate sulfatase (IDS). The IDS enzyme promotes the degradation of complex molecules named glycosoamminoglycans (GAGs). In particular IDS promotes the degradation of dermatan sulfate and heparan sulfate, which once degraded can be eliminated from the cells. In Hunter Syndrome these compounds accumulate as un-degraded molecules in the cells of the patients.
The cells, overloaded with these molecules within particular organelles, the lysosomes, initiate a systemic degeneration of all of the tissues of the body. To date, there is no effective treatment for MPSII and gene therapy is an attractive approach to cure the MPSII syndrome. To this aim we used the Hunter mouse model that exhibits many of the characteristics of MPSII, including skeleton abnormalities such as coarse faces, macrodactlya, and elevated accumulation of GAGs in the urine and many organs. We generated a viral vector carrying the human IDS that produces the active enzyme exclusively in the liver. The viral particles were administrated intravenously (IV) to group of adult MPS II mice and mainly targeted the liver. When the IDS enzyme was produced from the liver it was secreted into the bloodstream and taken up by the other organs leading to the correction of the systemic defects. We analyzed the correction of the disease phenotype in treated mice after one and seven months. In both groups, upon the gene therapy treatment, the plasma levels of IDS enzyme were increased in all treated mice and the accumulation of GAGs in the urines was reduced. We also carried out behavioral tests on the treated animals that performed as well as the normal wild-type control mice. Furthermore, even the skeletal abnormalities, such as coarse faces and macrodactlya, resulted completely corrected. The tissues of treated mice were also analyzed and showed a rescue of the enzymatic activity of IDS and complete GAGs clearance. Finally, microscopically, there was a marked reduction in vacuolization in all of the organs examined.
We also characterized the CNS defects of the MPSII mice and found degeneration of Purkinje cells in the cerebellum and GAGs accumulation in the choroid plexus. The described gene therapy approach has the caveat that it does not correct the central nervous system defects. The IDS secreted from the liver into the bloodstream does not cross the brain blood barrier and therefore does not reach the brain. To rescue the central nervous system phenotype, we are performing experiments based on the delivery of the IDS adeno-associated vector in the cerebrospinal fluid and direct injection in the cerebellum. Preliminary results showed a partial clearance of the accumulation of GAGs in the choroid plexus. The results achieved so far demonstrate possibility and the efficacy of gene therapy by AAV carrying the active IDS to treat mucopolysaccharidosis type II.?
Washington University, St. Louis, MO
?Characterization of the Systemic Inflammatory Response to Lysosomal Storage?
Specific Aim 1: The goal of this Specific Aim was to determine if the abnormal clinical signs observed in the MPS I mouse (adipose storage deficiency, anemia, muscle wasting) were common to other lysosomal storage diseases.
As presented in the preliminary data section of the proposal every mouse model of lysosomal storage disease had a significant decrease in adiposity (15-57% decrease compared to normal control animals). With respect to anemia, the models of MPS (I, IIIB and VII) all had anemia as presented in the preliminary data. However, neither the Neimann-Pick AB (NPAB) nor the infantile neuronal ceroid lipofuscinosis (INCL) models had significantly decreased hematocrits. We are currently sectioning quadriceps muscles from all of the models to determine if there is significant muscle wasting.
Specific Aim 2: The goal of this Specific Aim was to determine if the abnormal levels of pro- inflammatory molecules seen in the MPS I model were common to other models of lysosomal storage disease.
Our preliminary data showed that both MPS I and NPAB mice had significantly elevated levels of several cytokines and chemokines. Interestingly, the biggest changes were in the levels of circulating chemokines and soluble VCAM. We have completed our analyses of the MPSIIIB, MPS VII and INCL models. The levels of pro-inflammatory molecules varied among the various models but there was evidence of systemic inflammation in all of the models. MPS IIIB mice had elevated levels of MCP-1, MCP-3, MIP-1a, VCAM and IL-1b. MPS VII mice had elevated levels of MCP-1, MCP-3 and VCAM. INCL mice had elevated levels of soluble VCAM. Therefore, inflammation appears to be a common clinical feature of lysosomal storage diseases. Interestingly, MCP-1 and/or VCAM are elevated in every model of lysosomal storage disease.
Specific Aim 3: The goal of this Specific Aim was to test the hypothesis that systemic inflammation contributed to the disease progression in murine models of lysosomal storage diseases.
We proposed several different approaches to test this hypothesis. Since MCP-1 was significantly elevated in nearly every model of lysosomal storage disease, we obtained the MCP-1-null mouse from the Jackson Laboratory and have moved both the MPS I and NPAB mutations onto that genetic background. We currently have colonies of MCP-1- and NPAB-
deficient mice that carry both the disease mutation and the MCP-1 mutation and are generating double mutant animals to be tested for adiposity, anemia and muscle wasting.
We have also created an HIV-based gene transfer vector encoding the M3 protein from g- Herpes virus. M3 is a secreted protein that binds most chemokines with high affinity and inactivates the pro-inflammatory function. In this way we can determine the effects of selectively inhibiting the function of the elevated chemokines present in the murine models. We currently have several MPS I and MPS IIIB mice that have been injected with the HIV-M3 virus and are generating additional animals to complete those experimental groups.
In a similar fashion, we have obtained a transgenic mouse model that expresses M3 from an inducible promoter in the presence of the antibiotic doxycycline. We have moved the transgenes onto the MPS I background and currently have mice that are expressing high levels of M3 systemically. All of the animals will be analyzed at 5 or 7 months of age to determine the effects of inhibiting a host of different chemokines (HIV-M3 and transgenic M3) or just MCP- 1’s actions (MCP-1 knockout).
In 2004 the National MPS Society awarded grants to Dr. Maria Pia Cosma for her work “Gene Therapy for MPS II” and to Dr. Mark Sands for his work, “Systemic inflammation associated with lysosomal storage diseases”. Below are the reviews they submitted summarizing the second year of their research.
The goal of this research was to determine the underlying cause of the anemia and adipose storage deficiency associated with MPS I and whether this phenotype was common to other lysosomal storage diseases (LSDs). We also proposed to determine if the systemic inflammation associated with MPS I was common to other LSDs and whether a reduction in the levels of certain pro-inflammatory molecules affected the disease progression. We initially showed that MPS I mice had a 41% decrease in body fat and an 18% decrease in hematocrit when compared to normal litter mates. The anemia was not a common finding among the other models of LSD. However, the inability to accumulate normal fat stores is common to other LSDs. Mouse models of MPS IIIB, MPS VII, Niemann-Pick AB (NPAB), and Infantile Neuronal Ceroid Lipofuscinosis (INCL) have 37, 51, 42, and 15% decreases in body fat, respectively, when compared to normal litter mates. We tested a number of simple hypotheses that might explain this decrease in fat stores. There was no significant difference in food intake, physical activity (open field test), metabolic rate (O2 consumption), fecal content (free fatty acids, triglycerides, cholesterol), serum content (free fatty acids, triglycerides, cholesterol), muscle triglycerides, or liver triglycerides between affected animals and normal controls. However, in the process of searching for the cause of the adipose storage deficiency, we discovered that the level of pro- inflammatory molecules (specifically chemokines) was significantly elevated in the MPS I mouse. We discovered similar changes in the other models of LSD. Interestingly, MCP-1, MCP-3 and soluble VCAM were elevated in the serum of every model we tested, and lymphotactin, M-CSF, MIP-1g, and TIMP-1 were elevated in 3 out of 5 of the models. There is growing evidence that chemokines can have a direct effect on adipocytes and fat metabolism. We hypothesized that inhibition or genetic deletion of certain chemokines would affect, perhaps ameliorate the adipose deficiency associated with the models of LSD. We created a mouse model of MPS I that also expressed the pan chemokine-binding protein M3. M3 is a g-herpes virus-encoded protein that binds and inactivates many chemokines. We also moved the MPS I and NPAB mutations onto a mouse model that is genetically deficient in MCP-1. We chose the knockout animal of MCP-1 since it is one of the chemokines that is elevated to the highest levels in every animal model of LSD. Neither the inhibition of the chemokines by M3 nor the genetic deletion of MCP-1 led to any increases in adiposity in either the MPS I or the NPAB mouse. Although our hypothesis that chronic inflammation associated with LSDs leads to adipose storage deficiency appears to be incorrect, these studies have enabled us to develop several additional hypotheses that could explain this phenotype. We are currently testing the hypotheses that either malabsorption or a change in the gut biota result in the adipose deficiency that is common to the mouse models of LSD. We believe that the adipose storage deficiency is an important clinical finding and correction of this phenotype might improve the quality of life of the affected children.
Dr. Maria Pia Cosma, TIGEM, Naples, Italy
Our results have been published in the following paper:
Cardone M, Polito VA, Pepe S, Mann L, D’Azzo A, Auricchio A, Ballabio A and Cosma MP.(2006). Correction of Hunter syndrome in the MPS II mouse model by AAV2/8-mediated gene delivery. Human Mol Genet, April,15(7), 1225-36 Mucopolysaccharidosis type II (MPS II; Hunter syndrome) is a lysosomal storage disorder caused by a deficiency in the enzyme iduronate 2-sulfatase (IDS). At present, the therapeutic approaches for MPS II are enzyme replacement therapy and bone marrow transplantation, although these therapies have some limitations. The availability of new AAV serotypes that display tissue-specific tropism and promote sustained expression of transgenes offers the possibility of AAV-mediated gene therapy for the systemic treatment of lysosomal diseases, including MPS II. We have characterized in detail the phenotype of IDS-deficient mice, a model of human MPS II. These mice display a progressive accumulation of glycosaminoglycans (GAGs) in many organs and excessive excretion of these compounds in their urine. Furthermore, they develop skeleton deformities, particularly of the craniofacial bones, and alopecia, they perform poorly in open-field tests and they have a severely compromised walking pattern. In addition, they present neuropathological defects. We identified loss of Purkinje cells and cellular vacuolization in different regions of the brain: the hippocampus, thalamus, cerebellum and brainstem. We have designed an efficient gene therapy approach for the treatment of these MPS II mice. AAV2/8TBG-IDS viral particles were administrated intravenously to adult MPS II mice. The plasma and tissue IDS activities were completely restored in all of the treated mice. This rescue of the enzymatic activity resulted in the full clearance of the accumulated GAGs in all of the tissues analyzed, the normalization of the GAG levels in the urine and the correction of the skeleton malformations and of the locomotor disabilities. Furthermore a partial clearance of the GAG accumulation was also evident within the choroid plexus in the treated mice. This was surprising given the presence of the hemato- encephalic barrier. We predict that due to the very high levels of IDS in the plasma, which ranged from 16 to 70 times higher than the normal wild-type values, a fractional amount of the enzyme crossed the barrier and corrected the defect. Overall, our findings suggest that this in vivo gene transfer approach has potential for the systemic treatment of patients with Hunter syndrome.
The National MPS Society awarded seven research grants for 2003. Each grant is for
$30,000/year for two years. Funding for the grants was provided by the generous donations of our members for syndrome specific research and also by the money raised by the 2002 walk/runs. We are very grateful to the R. A. Bryan Foundation for funding one of the MPS III grants. Twenty-one proposals were received, including 6 for MPS I and 12 for MPS III. We regret that no proposals were received for MPS II, so that grant will not be funded in 2003.
Members of our Scientific Advisory Board reviewed the proposals; the final determination of funding was made by the Board of Directors.
Dr. Elena Aronovich, Pediatrics and Institute of Human Genetics, University of Minnesota, Minneapolis, MN
The Sleeping Beauty transposon, a novel, non-viral integrating system for gene transfer, will be tested as a tool for gene therapy of mucopolysaccharidosis type I. Our preliminary data obtained with an MPS VII mouse model indicate the feasibility of this approach. Using the transposon for expression of b-glucuronidase, we achieved enzyme levels that were sufficient to reverse lysosomal pathology in adult mouse livers in a 2-month experiment. Here, we propose to construct therapeutic transposon vectors carrying a human a-L-iduronidase gene that will be injected intravenously into MPS I mice. Correction of a-L-iduronidase enzyme deficiency and lysosomal pathology will be evaluated in mouse livers in a long-term (4 months) study. The biodistribution of the therapeutic transposon will be also determined. The proposed experiments will lay groundwork for future application of SB-mediated gene transfer to treatment of MPS diseases in human patients.
Dr. Judith Ogilvie, Ophthalmology & Visual Services, Washington University, St. Louis, MO
Mucopolysaccharidoses (MPS) are progressive diseases usually resulting from genetic defects in one of the lysosomal enzymes. Treating the eyes of adult MPS VII mice with virus-mediated gene therapy reduces lysosomal storage in treated eyes and in some parts of the brain. However, the severe systemic disease complicates accurate functional testing. Determining how much function can be recovered after treating established lysosomal storage in the nervous system is important, because most individuals with these diseases are diagnosed only after they begin to
show symptoms. This proposal’s goals are to examine whether intravitreal gene therapy improves vision function in MPS IIIB mice, and to lay the groundwork for future studies testing higher brain function. These mice have well-characterized pathology in their eyes and brains. Their longer life-span allows us to follow expression of the therapeutic enzyme longer than in MPS VII mice. Furthermore, this study will show whether transport from the eye into the brain works with a different lysosomal enzyme. Finally, since MPS III diseases predominantly affect the nervous system with mild systemic complications, behavioral tests can be performed at later ages. The proposed studies will provide the necessary preliminary information for designing cognitive function experiments in MPS IIIB mice.
Dr. Philippe Moullier and Matthew Ellinwood, Laboratoire de Therapie Genique, Nantes Cedex, France
The focus of this work is the further development of the canine model of MPS IIIB with an emphasis on gene therapy. 1) We propose to treat affected dogs at birth intravenously with gene therapy vectors to evaluate the ability of the normal gene to produce enzyme that is capable of being secreted into the blood and cross correct cells in other parts of the body. While this approach may not directly address the serious issues of brain disease in MPS IIIB, it provides important information about whether the gene is capable of producing normal enzyme that can be taken up by other cells. This is critical information, necessary for effective gene therapy to treat the brain disease seen in MPS IIIB. 2) We will evaluate gene therapy to the brain, via the direct injection of gene therapy vectors or stem cells. 3) Concurrent with these projects will be the continued study of the canine model, with an emphasis on identifying signs of disease that will help evaluation therapy.
Dr. Sharon Byers, Women’s and Children’s Hospital, North Adelaide, Australia
MPS disorders arise from a deficiency in an enzyme required for the degradation of complex carbohydrate molecules. Children with MPS display symptoms that include but are not limited to reduced height, blindness and brain pathology. Multi-tissue treatment for MPS is the subject of intensive research and centres around increasing the amount of deficient enzyme through enzyme replacement therapy (ERT), bone marrow transplantation or gene therapy. ERT in particular has shown great promise and has reached clinical trial for several MPS. However, some tissues with limited access to the circulation are not amenable to intravenous ERT. These include the brain, cornea and cartilage. Current therapy protocols thus do not address all sites of pathology. In this project therapy will be approached from a different direction. The synthesis of the complex
carbohydrate substrate normally degraded by the deficient enzyme will be reduced to a level that more closely matches the reduced enzyme activity observed in MPS patients, this concept is termed substrate deprivation therapy. Small sugar analogues of glucose and galactose will be synthesized and assessed for their ability to inhibit carbohydrate synthesis. The advantage of this type of therapy is that the inhibitors will equilibrate with cellular pools of carbohydrate throughout the body. All sites of pathology in MPS are thus amenable to substrate deprivation therapy.
Dr. Calogera Simonaro, Mr. Sinai School of Medicine, New York, NY
This grant proposal will study joint and bone disease in animals with mucopolysaccharidosis (MPS) type VI (Maroteaux-Lamy disease). Our goal is to gain a better understanding of this disorder and to develop new and/or improved therapies. We will specifically study the mechanism(s) causing cell death in cartilage and bone, and the role of inflammation in the disease process. We will also examine enzyme therapy and other therapeutic strategies for these conditions. It is our hope that these studies on animal models of MPS VI will lead to more effective therapy for this and other MPS disorders, particularly in the joints and bones.
Dr. Urs Giger, Medical Genetics, University of Pennsylvania, Philadelphia, PA
Mucolipidosis II (ML II), also called I-cell disease, is a unique cellular storage disease leading to severe skeletal malformations, growth and mental retardation, and death within the first decade of life. Although ML II is caused by faulty trafficing of enzymes to reach cellular organelles (lysosomes), it shares many clinical features of the more common forms of mucopolysaccharidoses (MPS). We have established a colony of domestic shorthair cats with naturally-occurring ML II, the first model in which to study this rare storage disease. After we documented the clinical features in cats and mode of inheritance, we propose now to characterize the pathology of ML II in tissues from affected cats and compare the results to the scant information from human patients. Although the deficient enzyme has been identified, the molecular basis remains unknown in affected cats as well as humans. We therefore also propose to characterize feline I-cell disease at the molecular genetic level by sequencing the gene of the normal enzyme and identify the disease-causing mutation in affected cats. Thereby, the knowledge gained in feline ML II will likely further our understanding of this disease in humans and provides the necessary characterization for this animal model to become useful in the development and assessment of the safety and efficacy of novel therapeutic interventions.
Occasionally the MPS Sociey receives requests for grant funding outside our normal funding cycle. The year we received 2 excellent proposals, and the Board of Directors chose to award a one year grant of $8750 to Dr. Elsa Shapiro at the University of Minnesota for the fellowship of Dr. Kendra Bjoraker. This grant was possible because not all the money allocated for the Partnership Grant was requested.
“Research Training in Psychosocial Development and Quality of Life in MPS Disorders”. Dr. Elsa Shapiro, University of Minnesota, Minneapolis, MN Few psychologists have expertise in MPS disorders. As a result, little research has been done on the factors that contribute to quality- of-life and psychosocial outcomes of children with MPS. We need to determine how new treatments (such as enzyme replacement or gene therapies) as well as established therapies such as bone marrow and cord blood transplant might alter quality-of-life and psychosocial status as well as the previously studied cognitive and language development. Also, this will enable us to find ways to improve the quality-of-life and psychosocial outcomes of these children.
We propose to develop a training program at the University of Minnesota to train a neuropsychologist to carry out such research to increase understanding of MPS. We have developed a detailed program curriculum. We are uniquely able to carry out training at Minnesota due to the number of physicians and psychologists here who can provide expert mentorship. In addition to training in the disciplines necessary to carry out this research, the trainee would carry out such a study during the year’s work. We hope that training such a person would be a model for other centers to pursue such research and training and would lead to federal funding.
The Board of Directors of The National MPS Society approved a 2003 Partnership Grant with Julia’s Hope Medical Research Foundation for Sanfilippo Syndrome. A total of 3 grant proposals were received, all of which were for MPS III research. The funding is for one year, with both groups granting $41,000.
Dr. John Hopwood, Lysosomal Diseases Research Unit, North Adelaide, Australia.
This grant is a Partnership Grant with Julia’s Hope Medical Research Foundation for Sanfilippo Syndrome. The funding is for one year with both groups granting $41,000. The aims of this research project are to utilize a unique and recently described non-viral transposon system (named Sleeping Beauty [SB]) to facilitate gene augmentation therapy in lysosomal storage diseases (LSD) that affect the brain, with the view to preventing or reversing clinical signs and symptoms. We will begin by testing our potential new therapy in the mouse model of MPS III-A. This research will determine the breadth of the effect of the treatment on the brain. As LSD affect widespread areas within the brain, our treatment needs to have access to most of the brain to be effective in reversing the clinical signs and symptoms. The research will also determine whether the clinical manifestations of these disorders can be prevented in newborn animals, delayed/reversed in young animals or reversed in older animals
Pediatrics and Institute of Human Genetics University of Minnesota, Minneapolis, MN
“SB (Sleeping Beauty) Transposon-mediated Gene Therapy for MPS I”.
The Sleeping Beauty (SB) transposon system created at the University of Minnesota is one of the few non-viral gene therapy systems that are able to integrate genes into human chromosomes.
This is especially relevant for treating genetic disorders, such as mucopolysaccharidoses, that require life-long expression of the therapeutic gene. The purpose of this funded project is to see if the SB transposon system can efficiently deliver the ?-L-iduronidase (IDUA) gene to chromosomes in MPS I mice for long-term correction of the disease.
The SB transposon system consists of two parts: the transposon that carries the therapeutic gene and the source of transposase enzyme that cuts the gene out of the plasmid and pastes it into the chromosomal DNA. Without the transposase, the therapeutic gene will still be expressed, but only short-term, since it is unlikely that it will be integrated into the chromosome. We constructed a series of SB transposon vectors that all provided high IDUA activity but differed in the level of the transposase enzyme. The constructs were tested for transposition efficiency in cell culture. The most efficient construct was then injected into MPS I mice that were completely deficient in IDUA activity. The control group of MPS I mice did not get the transposase. The mice were immune-suppressed with the drug cyclophosphamide to allow detection of IDUA, which is recognized as a foreign protein in the MPS I mice. Two other groups of MPS I mice were treated with the therapeutic transposon vectors, but without immune suppression. Blood samples were collected one day after treatment and once every two weeks thereafter. Delivery of the therapeutic transposon was considered technically successful by the observation of plasma IDUA activity >100-fold of wild type levels the day after treatment. In treated MPS I mice that did not receive cyclophosphamide, IDUA activity was undetectable in all mice six weeks after delivery of the therapeutic gene. No IDUA activity was detected in the livers of these mice three months after plasmid administration. In all immune-suppressed mice, the initial plasma activity levels dropped approximately 150-fold over the first two weeks after delivery, but then remained stable in transposase-positive mice (up to five times higher than in the wild-type mice). But in transposase-negative (control) MPS I mice the IDUA levels continued to decline gradually over three months. In the liver, at three months, IDUA activity was detected in both transposase- positive and transposase-negative groups. However, in the former group, these levels were on average four fold higher. The obtained levels of IDUA activity were sufficient to reduce dramatically the number and size of pathologic inclusions in liver, up to their elimination, which was demonstrated by staining with toluidine blue of liver sections.
Thus, in a long-term experiment (three month experiment) a single dose of SB transposon system resulted in partial to complete correction of IDUA activity in livers of treated adult MPS I mice.
Our future efforts will be to: conduct a longer-term (6-12 month) study of IDUA expression in treated mice, define a pattern of biodistribution of the circulating IDUA enzyme in the treated MPS I mouse organs, and find a way to prevent more efficiently the inhibitory antibody response to the therapeutic IDUA enzyme.
The results of this year’s work have been reported at the annual WORLD Lysosomal Disease Research Network Symposium, Minneapolis, Minnesota, May 13-15, 2004:
Washington University School of Medicine, St. Louis, MO “Intravitreal gene therapy in MPS IIIB knockout mice”
We have made considerable progress on two goals. First, we have successfully built a breeding colony of MPS IIIB knockout mice. These animals are now available in sufficient numbers for experimental testing.
Secondly, we are making the gene transfer vector. Although Fu et al (Mol Ther 5:42-49, 2002) have constructed rAAV vectors encoding NAGLU cDNAs, we decided to prepare our own vector in order to be able to compare results from this study to those previously reported for AAV-GUSB in MPS VII mice (Hennig et al, J Neurosci, 23:3302, 2003; Hennig et al, Mol Ther, in press). We started with the same backbone previously used for a ?-glucuronidase vector. We then inserted human NAGLU cDNA from pCMV-hNAGLU (a kind gift from Elizabeth Neufeld), excised with EcoRI and ligated into pTR-CAGG?530 in place of GUSB (excised by partial digest). The insert orientation was confirmed by restriction digest as follows:
| ENZYME | PREDICTED SIZES | BANDS SEEN |
| EcoRI | 4829, 2481, 1590 bp | 5.0, 2.3, 1.6 kB |
| Sma I | 4540, (3221+1390) or (3686+1189) | 4.5, 3, ~1.5 kB |
| Bgl II | 4940, (3586+370) or (2286+1515) | 5.0, 3.5 |
Sma I+Not I 4540, (2888+1385) or (3514+760), 172
These results indicate no recombination of inverted terminal repeats and that the NAGLU cDNA is in the correct orientation to produce protein.
In order to determine whether the construct produces active NAGLU enzyme, we transfected four fibroblast cell lines prepared from NAGLU -/- and +/+ littermate tissues according to the Mark Sands’ Lab protocol. We tested the following conditions: transfection with 3 dilutions of plasmid containing the AAV genomic construct, control plasmid DNA, medium with no DNA, and not transfected. NAGLU -/- cell cultures showed a slight but clear, dose-dependent increase in positively stained cells in wells that had been transfected with AAV-NAGLU construct.
NAGLU enzyme activity levels in medium from transfected cultures were slightly higher than untreated or sham-transfected culture medium for NAGLU-deficient flat cell cultures.
These results indicate that the construct does produce active enzyme and we are now proceeding to make the virus for gene therapy in the MPS IIIB knockout mouse.
Inserm U 0649-Laboratoire de Th?rapie G?nique, CHU H?tel-Dieu. Nantes,
France
“Evaluation of Gene Therapy in the Canine Model of MPS IIIB”.
The focus of this grant is the further development of the canine model of MPS IIIB with an emphasis on gene therapy. Broadly speaking there have been two main goals in this grant. The first group of aims involves the construction and analysis of gene therapy vectors to be used in vivo in the canine model that are designed to deliver normal copies of the canine N-acetyl-?-D- glucosaminidase (NaGlu) cDNA. The second group of aims involves further characterization of the canine model so that therapies can be evaluated in a timely manner, obviating the need to assess their impact on clinical signs, which are adult onset in this canine model.
In pursuit of the first set of aims, work is underway on the construction of a gene therapy vector containing the canine NaGlu cDNA in the context of a recombinant adeno-associated viral (rAAV) vector derived from the AAV serotype 2. This vector will be assessed by intracerebral injections in the canine model. In support of the second aim of the grant, work has progressed on a better understanding of the natural history of the disease in the canine model. The canine model of MPS IIIB is unlike other models in two respects. First, combined with the canine models of MPS IIIA, it is among the only large animal models of MPS disorders to show overt neurological clinical signs. Secondly, unlike the clinical signs of MPS seen in other large animal models, these clinical signs are of early adult onset. This latter fact makes it critical that biochemical markers and/or pathological findings which differ between normal and affected dog be identified at a young age. To this end we have pursued analysis of affected dogs looking at histopathological signs of disease, as well as at biochemical changes in the brains of affected dogs. Histological, lesions associated with lysosomal storage can be distinguished in the liver
and kidney of affected dogs as early as 3 months of age. These findings were also seen in affected dogs at six months of age. No findings of lysosomal storage has been seen in the central nervous system of affected dogs at these ages using convention histopathological techniques, however semi-thin section analysis will be pursued. In an effort to find central nervous system biochemical markers associated with disease in the canine model, we have conducted ganglioside analysis of the cerebral gray matter of affected dogs, ages three months to six years of age. This work was done in at the Lyon-Sud Medical School in Lyon, France, in collaboration with Dr Marie T. Vanier and with the assistance of her graduate student Lucie Verot.
Gangliosides, some types of which have long been known to accumulate in the brains of patients with some forms of the MPS disorders, were found to be elevated in dogs as early as 3 months of age. The accumulated gangliosides (GM2 and GM3), remain elevated relative to age matched normal controls from 3 months of age onward, and this elevation increases until the end stages of the disease.
In the course of this year of the grant, Dr. N. Matthew Ellinwood, who was funded as a post- doctoral fellow by this grant, was selected to begin an assistant professorship in the Animal Genetics group within the Animal Science Department at Iowa State University, in Ames Iowa. This is primarily a research appointment, and Dr. Ellinwood, whose position begins October 1, 2004, will continue to work on canine MPS IIIB as the major focus of his research. The canine breeding colony, the establishment of which was supported by the National MPS Society, has been housed at the University of Pennsylvania, and is in the process of being transferred to Iowa State University, a process which will be completed this fall. In consideration of Dr. Ellinwood’s change of status, it has been proposed to the National MPS Society that the second year of this grant be transferred to Dr. Ellinwood at Iowa State University, where it will serve as the source of a stipend for a post-doctoral fellow in Dr. Ellinwood’s research laboratory.
Findings presented above have been presented in abstract form at an NIH symposium on the Glycoproteinoses and Related Disorders (April 2004), and are submitted to the American Society of Human Genetics meeting (October, 2004). All work present has acknowledged the funding support of the National MPS Society.
Department of Genetic Medicine,
Women’s and Children’s Hospital, North. Adelaide, South Australia
“Inhibition of glycosaminoglycan synthesis as a therapy for mucopolysaccharidosis type IVA and VI”
The goal of this proposal is to develop and evaluate substrate deprivation therapy for MPS IVA and MPS VI, using small molecular weight inhibitors of glycosaminoglycan synthesis. In the first instance therapy for MPS IVA has been prioritized. One of the obstacles to evaluation of any type of therapy for MPS IVA is the availability of a convenient in vitro system to monitor correction of storage. The most widely used cell type, the skin fibroblast, does not synthesize or store significant amounts of keratan sulphate and is therefore not suitable for testing therapy regimens. Our first aim was to develop an in vitro assay system. To achieve this, the keratan sulphate containing domain (G1-G2) of the large cartilage proteoglycan, aggrecan, was isolated and cloned into 2 different expression vectors; an HIV-1 based lentivirus (pHIVmpsvG1-G2) and pCDNA3.1v8HisTOPO (pTOPOG1-G2). Normal cells infected with pHIVmpsvG1-G2 expressed low levels of the keratan sulphate domain as determined by Western blot. Work is in progress to optimize expression from both constructs and assess both skin fibroblast and bone osteoblast cells as mediators of expression.
The concept of substrate deprivation therapy and its application to keratan sulphate synthesis has been demonstrated in normal bovine articular cartilage cell cultures. Cartilage chondrocytes produce large amounts of keratan sulphate containing proteoglycans. The addition of either a general inhibitor of glycosaminoglycan synthesis or a small molecular weight inhibitor of keratan sulphate synthesis to cell culture medium resulted in a decrease in the level of keratan sulphate produced. Based on other experiments with different glycosaminoglycan types, we have shown that inhibition of glycosaminoglycan synthesis results in decreased storage of gag degradation products in the appropriate MPS skin fibroblast cells. Similar experiments will be performed with the keratan sulphate inhibitors once we have fully developed our in vitro assay for keratan sulphate storage. Large scale synthesis of the small molecular weight inhibitor of keratan sulphate synthesis has been initiated.
Department of Human Genetics, Mount Sinai School of Medicine, New York, NY “Joint & Bone Disease in the Mucopolysaccharidoses: Identification of New Therapeutic Targets & BioMarkers Using Animal Models”
The mucopolysaccharidoses (MPS) are inherited metabolic disorders resulting from the defective catabolism of glycosaminoglycans (GAGs). We previously used MPS animal models to investigate the pathophysiology of the joints and bones, major sites of pathology in these disorders, and found enhanced chondrocyte apoptosis and nitric oxide production associated with TNF-? and Il-1. We now report that the stimulation of MPS connective tissue cells by these inflammatory cytokines causes enhanced secretion of several matrix-degrading metalloproteinases (MMPs). In addition, expression of tissue inhibitor of metalloproteinase (TIMP)-1 was elevated, consistent with the enhanced MMP activity. These findings were not restricted to one particular MPS disorder or species, and are consistent with previous observations in animal models with chemically induced arthritis. BrdU incorporation studies also revealed that MPS chondrocytes proliferated up to five-fold faster than normal chondrocytes, and released elevated levels of TGF to counteract the marked chondrocyte apoptosis and matrix degradation associated with MMP expression. However, despite this compensatory mechanism, studies of endochondral ossification revealed a reduction in chondrodifferentiation in the growth plates. Thus, although MPS chondrocytes grew faster, most of the newly formed cells were immature and could not mineralize into bone. Our studies suggest that altered MMP expression, most likely stimulated by inflammatory cytokines and nitric oxide, is an important feature of the MPS disorders. These data also identify several proinflammatory cytokines, nitric oxide, and MMPs as novel therapeutic targets and/or biomarkers of MPS joint and bone disease. This information should aid in the evaluation of existing therapies for these disorders, such as enzyme replacement therapy (ERT) and bone marrow transplantation (BMT), and may lead to the development of new therapeutic approaches.
Urs Giger, PD Dr. med. vet. FVH
Section of Medical Genetics, University of Pennsylvania Philadelphia, PA
“Pathological and Molecular Characterization of Feline Mucolipidosis II: The First Model of Human I-Cell Disease”
Mucolipidosis II (ML II), also called I-cell disease, is a unique cellular storage disease leading to severe skeletal malformations, growth and mental retardation, and death within the first decade of life. Although ML II is caused by faulty trafficking of enzymes to reach cellular organelles (lysosomes), it shares many clinical features of the more common forms of mucopolysaccharidoses (MPS). We have established a colony of domestic shorthair cats with naturally-occurring ML II, the first model in which to study this rare storage disease.
Recently we have documented the clinical features in cats and the autosomal recessive mode of inheritance (Mazrier et al J Heredity 2003) and documented the close homology and minor differences between the disorder in humans and cats. As the pathology is hardly described in humans with ML-II, we were keen to characterize the pathology of feline ML II in tissues from affected cats and compare the results to the scant information from human patients. We have prepared histological preparations from autopsied animals and are analyzing each tissue by Jessica Caverly VMD PhD, a veterinary pathologist who received a Reentry Fellowship from NIH for these studies. Furthermore, we are extracting the various tissues to identify the specific storage material including mucopolysaccharides and gangliosides in collaborations with others. Similarly we have cultured fibroblast from affected cats to further characterize the specific inclusions so classic for I-cell disease. Finally, tissues with specific pathology will also be assessed by electron microscopy.
Although the deficient enzyme has recently been identified in humans, the molecular basis remains unknown in affected cats as well as humans. Through William Canfield at Genzyme we were able to gain access to the sequence of the human enzyme N-acetylglucosamine-1- phosphotransferase, compared that sequence to the recently released canine transferase sequence and have developed primers to amplify the exons from normal and affected cats. We have thus
far about 1kb amplified and sequenced and there seems to be close homology between species. We are also using fibroblast cultures for RT-PCR of the feline cDNA and thereby should be able to get the entire sequence shortly, therefore we are in a good position to characterize feline I-cell disease at the molecular genetic level.
Some of our initial findings were presented at the National MPS Society meeting in Mainz and further collaborations for storage pathology and biochemistry were established. Thereby, the knowledge gained in feline ML II will likely further our understanding of this disease in humans and provides the necessary characterization for this animal model to become useful in the development and assessment of the safety and efficacy of novel therapeutic interventions.
Inserm U 0649-Laboratoire de Th?rapie G?nique, CHU H?tel-Dieu. Nantes,
France
“Evaluation of Gene Therapy in the Canine Model of MPS IIIB”.
The focus of this grant is the further development of the canine model of MPS IIIB with an emphasis on gene therapy. Broadly speaking there have been two main goals in this grant. The first group of aims involves the construction and analysis of gene therapy vectors to be used in vivo in the canine model that are designed to deliver normal copies of the canine N-acetyl-?-D- glucosaminidase (NaGlu) cDNA. The second group of aims involves further characterization of the canine model so that therapies can be evaluated in a timely manner, obviating the need to assess their impact on clinical signs, which are adult onset in this canine model.
Lysosomal Diseases Research Unit, Women’s and Children’s Hospital North Adelaide, South Australia, Australia
The Sleeping Beauty transgene vehicle ? a potential new therapy for MPS-IIIA
The present study is investigating a potential new treatment for lysosomal storage disorders (LSD) that affect the brain, the ‘Sleeping Beauty transgene vehicle’. Sleeping Beauty (SB) is able to transport genetic material into cells. We believe that we will be able to use Sleeping Beauty to transport the genetic material into cells that is required to make lysosomal enzymes. In LSD patients, this would mean that cells treated with SB would make lysosomal enzyme ‘normally’, and these disorders could thereby be treated. In particular, we are excited about the ability of Sleeping Beauty to enter the cells in the brain. The MPS IIIA mouse model is being used in these studies.
Our preliminary studies have focused on the use of SB that has a red fluorescent tag (so that it can be located within the brain) (SB-dsRed), which has been constructed by Professor Clifford Steer and Dr Betsy Kren and colleagues. SB-dsRed has been used in the first instance to obtain some understanding of how SB moves around the brain from the injection site and how many cells it treats ? we can determine this by looking to see how many red cells there are in a section of mouse brain. Ideally, we would like SB to treat every cell within the brain. In the next phase of the study, we will utilise the SB vector that is capable of transporting the genetic material into cells that is required to make the lysosomal enzyme sulphamidase. SB-sulphamidase is being constructed by Dr Kren in Minnesota at present.
We have used the SB-dsRed vector and have undertaken: (1) preliminary studies investigating the potential of Sleeping Beauty as a future long-term treatment for LSD that affect the brain. The first step in this process has been to determine how long SB-treated cells are capable of producing the red fluorescent tag (or in the future, sulphamidase). We would like SB to enable cells to produce lysosomal enzymes on a long-term (years) basis, so that this treatment does not have to be given too frequently. These experiments have been carried out in human unaffected and MPS-IIIA cells grown in culture in the laboratory; and (2) subsequent studies where we injected SB-dsRed directly into the newborn and adult mouse brain to determine where the fluorescent tag is seen within the brain. The findings from this work are discussed below.
In more recent studies, newborn MPS IIIA mice received injections of SB-dsRed into the ventricles (fluid filled spaces) in the brain and the location of SB-treated cells (as determined by the presence of the red fluorescent protein) was determined two-weeks later. Treated cells were observed in even more widespread areas of brain. These experiments are continuing.
In summary, these preliminary studies pave the way for planned experiments using the SB- sulphamidase vector in MPS-IIIA mice (as a precursor to MPS-IIIA dog and then MPS-IIIA human studies). We are hopeful that cells in widespread areas of the brain will be treated with SB, thus alleviating the deleterious effects of reduced lysosomal enzyme activity in this disorder.
Dr. Judith Mosinger Ogilvie, Department of Biology, St. Louis University
The objectives of this project were to fully characterize the functional and pathological degeneration of the MPS IIIB mouse model of Sanfilippo Syndrome type B, to determine whether visual function could be restored through intravitreal gene therapy, and to lay the groundwork for rational design of studies to assess function in the central nervous system. Preliminary studies in MPS VII mice with virus-mediated gene therapy resulted in correction of lysosomal storage in treated eyes and in several areas of the brain. However, because of the severity of the disease, we could not test the long-term functional consequences of treatment in these mice.
We have succeeded in achieving the first objective with full characterization of the functional and pathological degeneration of the MPS IIIB mouse. Functional evaluation was performed with electroretinograms (ERGs) of retinas from MPS IIIB mice at 4, 8, 12, 16, 20, 30, 34, and 45 weeks of age. ERG testing of light- and dark-adapted mice produces a characteristic wave-form that allows for identification of deficits in different cell types within the retina. The dark-adapted retinal ERG response is depressed by 5 weeks and becomes progressively less sensitive with increasing age. The diminished sensitivity reflects a loss of rod photoreceptor function that achieves persistent significance after 15 weeks. No significant differences were observed at any age in retinal function after light adaptation indicating a normal level of cone photoreceptor function.
We have performed histological analysis on retinas from MPS IIIB mice at the same time points. At 4 weeks of age, the mutant retina appeared grossly normal, although localized abnormalities were seen in retinal pigment epithelium (RPE) cell shape consistent with loss of cell polarization and/or delamination. By 8 weeks of age, lysosomal storage could be seen in vascular cells and microglia in the inner retina. Occasional pyknotic nuclei could be seen in the outer nuclear layer, which is comprised of photoreceptor cells, and further disruption in the RPE was seen.
Photoreceptor degeneration, with shortening of the outer segments and cell loss in the outer nuclear layer, became apparent around 16 weeks with a decrease of 2-4 rows of nuclei by 20 weeks of age. Macrophage-like cells were apparent in the subretinal space between the RPE and photoreceptor outer segments by 16 weeks. Nearly half of the photoreceptor cells were gone by
30 – 34 weeks with only 3-4 rows of nuclei remaining at 45 weeks. Outer segments were further shortened and appeared swollen. Large, round, dense melanosome-like structures were seen in the RPE of mutant retinas, first becoming noticeable in the periphery of the mutant eye as early as 4 weeks, distributed throughout the RPE by 16 weeks, and becoming larger and more prominent by 30 weeks. In mutant retinas, unlike wild type controls, these structures were prominent in the mid to basal cytoplasm and were distinguished by their round shape.
This thorough characterization of the retina of the MPS IIIB mouse model of Sanfilippo Syndrome type B is an important step forward in investigating potential therapeutic interventions for this disease. These results have been presented in abstract form at the Association for Research in Vision and Ophthalmology meeting (Hennig, et al., 2006) and are currently in preparation for publication with additional characterization performed by Dr. Mark Sands and collaborators (Heldermon, et al).
Considerable effort was placed in producing a gene transfer vector. Initial results indicated that our construct produced active enzyme. MPS IIIB knockout mice and wild type controls were injected intravitreally with the vector and functional tests were performed at three time points. Tissue was harvested and processed. Unfortunately, the viral construct did not produce sufficient expression of the NAGLU enzyme to determine whether visual function could be restored through intravitreal gene therapy. Although we were disappointed with this result, other experiments performed concurrently with this work have enabled progress on the third objective. We collaborated with Drs. Mark Sands and Megan Griffey on the ppt1 mouse model of Batten’s Disease. Those studies (Griffey, et al., 2005) were successful in establishing the groundwork for the future design of studies to assess the ability of intravitreal gene therapy to improve CNS function in lysosomal storage diseases.
Publications:
Griffey, M., S.L. Macauley, J.M. Ogilvie, M.S. Sands. (2005) AAV2-mediated ocular gene therapy for infantile neuronal ceroid lipofuscinosis. Mol. Ther. 12:413-21.
Heldermon, C.D., Hennig, A., Vogler, C., Ohlemiller, K., Ogilvie, J.M., Breidenbach, A., Herzog, E.D., Sands, M.S. “Characterization of the murine model of Sanfilippo syndrome type B.” In preparation.
Hennig, A.K., M. Griffey, M.S. Sands, R.L. Gunkel, M.K. Murphy, J.M. Ogilvie. (2006) Electroretinogram Changes and Retinal Degeneration in Knockout Mouse Models of Four Lysosomal Storage Diseases. ARVO Abstract #5780 accessed at www.arvo.org. presented May 4, 2006 at the annual conference of the Annual Conference of the Association for Research in Vision and Ophthalmology.
University of Minnesota
The Sleeping Beauty (SB) transposon system created at the University of Minnesota is one of the few non-viral gene therapy systems that are able to integrate genes into human chromosomes to provide life-long expression of a therapeutic gene. The purpose of this funded project has been to see if the SB transposon system can efficiently deliver the a-L-iduronidase (IDUA) gene to chromosomes of liver in MPS I mice for long-term expression and correction of the disease. The SB transposon system consists of two parts, the transposon that carries the therapeutic gene and the source of transposase enzyme, which cuts the gene out of the plasmid and pastes it into chromosomal DNA. Without the transposase, the therapeutic gene can be expressed, but generally only short-term, presumably because as an unintegrated episome, it is either lost or recognized as foreign DNA and inactivated.
For this study we chose the very efficient, high-pressure “hydrodynamics-based” DNA injection, which targets the liver. We constructed SB transposon plasmid-based vectors and injected them into MPS I mice that were completely deficient of IDUA activity. Because our goal was in part to determine the efficacy of transposition as a way of providing long-term expression of IDUA protein, control groups of MPS I mice did not receive the transposase. Blood samples for plasma isolation were collected 1 day after treatment and once every 2 weeks thereafter. Plasma IDUA activity reached >100-fold of wild type levels on day 1 following treatment, but was essentially gone in all mice by 4-weeks. IDUA activity was not detected in the liver of mice 3 months after plasmid administration. We examined the duration of the transposon-delivered IDUA gene by PCR and IDUA expression in liver of unaffected mice over 6 months. As in the MPS I mice, plasma and liver IDUA activity reached supra-normal levels on day 1 and remained at this level for the first week, but reduced dramatically by the 2-week time point and by 4 weeks were indistinguishable from background. Notably, the presence of the IDUA transgene mirrored the IDUA activity time-line, i.e., the PCR product from the transgene was not detectable after 2 weeks post-injection. However, transposition was confirmed by an “excision assay” (which detects the PCR product of the plasmid that delivered the IDUA gene to the liver). The excision product was detectable for the first two weeks, but was undetectable thereafter. This suggests that cells that express the IDUA gene are cleared from the liver of treated mice by 4 weeks following injection. Our data suggest induction of an immune response either to the therapeutic protein or/and the cells that express the therapeutic gene.
This conclusion was supported by our observations that in all cyclophosphamide immune- suppressed MPS I mice, the initial 1-day plasma activity levels dropped approximately 150-fold by 2 weeks, but then persisted at detectable levels in some mice. IDUA levels were stable in transposase-positive mice (up to 5 times higher than in the wild-type mice) whereas in transposase-negative (control) MPS I mice IDUA levels declined over 3 months. In the liver, at 3 months, IDUA activity was detected in both transposase-positive and transposase-negative groups. However, in the former group, these levels were on average 4-fold higher. The obtained levels of IDUA activity were sufficient to dramatically reduce the number and size of pathologic inclusions in the liver as demonstrated by toluidine blue staining of liver sections. Thus, with immune suppression, a single dose of the SB transposon system resulted in partial to complete biochemical correction (cure) of IDUA activity in the liver of treated adult MPS I mice.
Our future effort will be directed at achieving systemic delivery of the therapeutic gene and finding a way to prevent immune responses to the therapeutic gene in MPS mice.
The results of this work have been reported in three platform presentations and one poster at international meetings:
American Society of Gene Therapy, 2005 Annual Meeting, St. Louis, MO, June 1-5, Duration of Expression of Sleeping Beauty Transposase by Hydrodynamic Injection of C57/BL6 Mice. Jason B. Bell, Elena L. Aronovich, Brenda Koniar Roland Gunther, Beth Larson- Debruzzi, Chester B. Whitley, R. Scott McIvor and Perry B. Hackett Mol. Therapy, 2005, v.11: S423
Third Annual International Conference on Transposition and Animal Biotechnology, Minneapolis, MN, June 23-24, 2005 : Long-term Expression of Sleeping Beauty Transposon in the Murine Models of Mucopolysaccharidosis (MPS) Type VII and Type I.
Elena L. Aronovich, Jason B. Bell, Lalitha R. Belur, Joel L. Frandsen, Roland Gunther, Brenda Koniar, David C.C. Erickson, John R. Ohlfest, R. Scott McIvor, Chester B. Whitley, and Perry
WORLD Lysosomal Disease Clinical Research Network Annual Symposium 2004, May 13- 15, Minneapolis, MN
The focus of this grant is the further development of the canine model of MPS IIIB with an emphasis on gene therapy. Broadly speaking there have been two main goals in this grant. The first group of aims involves the construction and analysis of gene therapy vectors to be used in vivo in the canine model that are designed to deliver normal copies of the canine N-acetyl-a-D- glucosaminidase (NaGlu) cDNA. The second group of aims involves further characterization of the canine model so that therapies can be evaluated in a timely manner, obviating the need to assess their impact on clinical signs, which are adult onset in this canine model.
In the course of this grant, originally awarded to the Laboratoire de Therapie Genique (Dr. Philippe Moullier), Dr. N. Matthew Ellinwood, who was funded from this grant as a post- doctoral fellow in Nantes, France, was selected to begin an assistant professorship in the Animal Genetics group within the Animal Science Department at Iowa State University, in Ames Iowa. This primary research appointment began October 1, 2004, and Dr. Ellinwood’s work will continue to focus on canine MPS IIIB. In consideration of Dr. Ellinwood’s change of status, it has been agreed that the second year of this grant be transferred to Dr. Ellinwood at Iowa State University. Furthermore, to ensure that the fellowship be used optimally, Dr. Ellinwood has requested an extension of the second year of the award, so that the fellowship is used for primary research after a suitable candidate can be identified and after Dr. Ellinwood’s laboratory and canine colony are settled at Iowa State University.
The canine MPS IIIB breeding and research colony was fully transferred from the University of Pennsylvania in December of 2004, and our first ISU litter was whelped in May 2005. Research collaborations are underway with both a veterinary clinical neurologist and radiologist at the School of Veterinary Medicine at ISU. These efforts will allow for gene therapy experiments and further characterization of the model. In addition the laboratory is now poised to begin gene therapy evaluations, and a post-doctoral fellow, Dr. Chun Coa, has been identified, and will begin work in August 1, 2005.
Work supported by this grant was presented at the American Society of Human Genetics meeting (October, 2004). All work present has acknowledged the funding support of the National MPS Society.
Dr. S. Byers, Women’s and Children’s Hospital, North Adelaide, South Australia
The goal of this proposal is to develop and evaluate substrate deprivation therapy (SDT) for MPS IVA and MPS VI. These MPS disorders arise from the deficiency of a lysosomal enzyme required for the degradation of keratan sulphate (KS) or dermatan sulphate (DS) glycosaminoglycan (gag) chains respectively. To be effective, SDT must therefore target the synthesis of these gag chains. By decreasing KS or DS synthesis, the balance between gag production and removal can be redressed in MPS IVA and MPS VI. Thus any remaining patient enzyme activity can more effectively degrade the reduced amount of gag arriving in the lysosome in the presence of inhibitor. We have synthesised a small molecular weight analogue of glucose and assessed its ability to inhibit the synthesis of KS and DS gags in cell culture and compared its effect with a non-specific gag synthesis inhibitor. Both inhibitors decrease gag synthesis in a dose dependent manner when added to the culture medium of normal bovine cartilage cells. Using a combination of size exclusion chromatography and enzyme digestion to identify individual gags, a >50% decrease in the synthesis of the KS-gag containing fraction but only a 15% decrease in the DS-gag containing fraction is observed, implying a differential effect on the two gag populations. Work is currently underway to characterize the size and structure of KS gags synthesised in the presence of inhibitor. These results offer “proof of concept” that SDT targeting gag synthesis as a treatment for MPS disorders has the potential to be a feasible therapy option.
Dr. Calogera Simonaro, Human Genetics, Mount Sinai School of Medicine
The overall goal of our research is to fill the void in our understanding of MPS bone and joint disease and to develop new and improved therapies that might benefit MPS patients. We specifically study two animal models with MPS VI, but anticipate that the results obtained can be applicable to the general class of MPS disorders and benefit a wide range of patients.
Our studies carried out over the past five years (funded in part by the MPS Society and published in two peer-reviewed papers) have revealed that glycosaminoglycan GAG accumulation is a direct cause of chondrocyte death in the articular cartilage and growth plates of MPS animals, leading to abnormal matrix homeostasis. This enhanced cell death also triggers a series of signaling events that lead to marked inflammatory disease. Together, these two factors (enhanced cell death and inflammation), lead to the characteristic bone and joint disease in the MPS disorders. In addition, cellular defects associated with the maturation of MPS growth plates are likely contributing to abnormal bone growth.
Enzyme replacement studies in MPS animals and human patients have revealed that chondrocytes in joints and bones are difficult to reach following injection due to the poor vascular supply to these tissues and the fact that the target cells are embedded in a dense, negatively charged matrix. With the availability of enzyme replacement it is important to continue to investigate new approaches for improving enzyme delivery to these critical target tissues. We have modified the charge on these enzymes to make them less negatively charged, so that they might penetrate the cartilage matrix more efficiently and enter chondrocytes. Our data supports the notion that altering the charge might enhance its therapeutic usefulness for cartilage and bone.
Our findings have important implications for the treatment of MPS individuals, as well as for the identification of novel biomarkers to monitor disease progression and therapeutic efficacy.
Urs Giger, University of Pennsylvania Research Services
Mucolipidosis II (ML II), also called I-cell disease, is a unique cellular storage disease leading to severe skeletal malformations, growth and mental retardation, and death within the first decade of life. Although ML II is caused by faulty trafficking of enzymes to reach cellular organelles
(lysosomes), it shares many clinical features of the more common forms of mucopolysaccharidoses (MPS). We have established a colony of domestic shorthair cats with naturally-occurring ML II, the first model in which to study this rare storage disease.
Recently we have documented the clinical features in cats and the autosomal recessive mode of inheritance (Mazrier et al J Heredity 2003) and documented the close homology and minor differences between the disorder in humans and cats. Clinical signs seem to be rapidly progressive with leg deformities evident from birth; furthermore these kittens also develop retinal and corneal changes that are being further defined. As the pathology is hardly described in humans with ML-II, we were keen to characterize the pathology of feline ML II in tissues from affected cats and compare the results to the scant information from human patients. We have analyzed histological preparations of various tissues from autopsied animals. Interestingly the storage lesions seem to develop slower and be restricted to specific tissues (Caverly et al in preparation). These tissues will be further assessed by electron microscopy. Similarly, specific storage material including mucopolysaccharides and gangliosides, extracted in collaborations with others, could only be found in a limited number of tissues. Finally, we have cultured fibroblast from affected cats to further characterize the specific inclusions so classic for I-cell disease.
Although the deficient enzyme has recently been identified in humans, the molecular basis remains unknown in affected cats as well as humans. Through William Canfield at Genzyme we were able to gain access to the sequence of the human enzyme N-acetylglucosamine-1- phosphotransferase (GNTPA), compared that sequence to the partial shotgun feline sequence of the transferase. We have completed the sequence of the entire feline GNTPA and characterized the close homology to the human GNTPA sequence and gene structure. Comparing the sequence of affected and normal healthy kittens we have identified a putative disease-causing missense mutation Tcherneva, Seng, Caverly, unpublished). Further studies are in progress to establish a screening test and to characterize the effect on the protein.
Some of our initial findings were presented at the National MPS Society meeting in Mainz and further collaborations for future collaborations have been established internationally. Thereby, the knowledge gained in feline ML II will likely further our understanding of this disease in humans and provides the necessary characterization for this animal model to become useful in the development and assessment of the safety and efficacy of novel therapeutic interventions.
2002 Research Grants
2002 Research Grant PDF Download
“GGA proteins and I-cell disease”
New diseases involving proteins that control vesicle traffic within cells are currently being recognized and many display a similar clinical presentation to that seen in lysosomal storage disorder patients. A new group of molecules called GGA proteins (Golgi localized, gamma ear containing, ARF binding proteins) have been shown to be involved in vesicle traffic within cells and interact with the mannose-6-phosphate receptors that normally target and transport lysosomal proteins to lysosomes. In this project, we planned to investigate patients characterized as having either I-cell disease or MLIII. Some of these patients will obviously have a deficiency in the enzyme N-acetylglucosamine 1-phosphotransferase that has been shown to cause I-cell disease. It is now recognized that mutations in the two separate subunits of this enzyme give rise to a similar syndrome. However, a number of patients with symptoms consistent with I-cell disease may also represent a disorder that has not been previously described. We hypothesized that a defect in one of the GGA proteins should give an identical clinical presentation to I-cell disease, based on the inability to correctly traffic mannose-6-phosphate receptors. We proposed two specific project initiatives: 1. Develop monoclonal antibodies to specific regions on GGA proteins to allow the study of the cell biology and intracellular traffic of normal and mutant GGA proteins. 2. The characterization of I-cell and MLIII patients to identify potential defects in GGA proteins and vesicle traffic.
We have generated a panel of monoclonal antibodies and a specific polyclonal antibody to each of the GGA proteins; GGA1, GGA2 and GGA3. The antibodies produced, show specific reactivity to the respective GGA proteins and at least one antibody from each set detected denatured protein and showed reactivity on Western blots. Several of the monoclonal antibodies have had the positional location of their antibody reactivity mapped onto the respective GGA protein (ie. epitope mapped). We have developed immune quantification assays for determining intracellular levels of GGA proteins in cell extracts and optimized each of the respective assays. We have analyzed a panel of fibroblast cells and shown similar total levels of each GGA protein in I-cell patient (n=8) and control cell (n=7) lines. We have also analyzed these cell lines by immunofluorescence using the GGA antibodies, together with a panel of antibodies to lysosomal membrane markers, to determine the intracellular distribution of these markers. The study suggests that the panel of I-cell lines investigated have different patterns of marker distribution to the normal controls. Moreover, within the I-cell patient group there were at least two distinct patterns of marker distribution. We are currently investigating this altered marker distribution in relation to patient severity and the molecular defect in each patient.
North Adelaide, South Australia , Australia “Enzyme therapy in the CNS of MPS IIIA Mice”
MPS IIIA results from the deficiency of the enzyme known as sulphamidase. The major hurdle in being able to treat MPS IIIA patients is enabling this enzyme to get into the brain (the central nervous system, or CNS) to reduce the amount of storage in brain tissue. In this project we used a MPS IIIA mouse model to evaluate enzyme replacement therapy (ERT) on the development of CNS by investigating:
We used a laboratory-made form of the human enzyme using a process known as recombination. The resulting enzyme is known as recombinant human sulphamidase (abbreviated to rhNS). The mice were separated into different groups and some were treated with enzyme while others were not. In one group, MPS IIIA mice were given an injection of enzyme into their temporal vein.
The enzyme doses used in this group were either 1 milligram of enzyme per kilogram of body weight (mg/kg) or 5 mg/kg rhNS on day 0 of life; in a second group MPS IIIA and unaffected mice were not given enzyme. All mice were given a 2nd intraperitoneal injection at one-week of age and a 3rd and final injection into their tail vein at two-weeks of age. All mice underwent testing to measure their ability to function at 13- and 15-weeks of age. One important measure of function we used was the gait width test, which tested the mouse’s ability to walk. Between 6-10 weeks of age MPS IIIA mice display a characteristic narrowing of gait width when compared to unaffected mice. Treatment with rhNS from birth normalized gait width at both ages tested. Both enzyme doses were equally effective.
Samples of mouse brain and liver were taken at different time points after injection to determine both the effect of the enzyme and its distribution in these tissues. We observed a reduction in the amount of storage in all brain regions studied except one, the olfactory bulb (a part of the brain that is involved in giving us our sense of smell) at four-weeks of age in both of the treated groups of mice. Once again, there was no difference between the 1 mg/kg and 5 mg/kg groups.
A constant problem in trying to obtain large numbers of MPS IIIA mice occurs because the mothers of MPS IIIA mice have a tendency to eat their young. In this study we mated female MPS IIIA mice treated with rhNS with untreated male MPS IIIA mice and found that enzyme replacement from birth improves the ability of MPS IIIA mice to maintain their young.
MPS IIIA mice have an average life span of 10-12 months. Ten-months after enzyme injections, all untreated MPS IIIA mice had either died or been euthanased due to health issues.
Importantly, at this age MPS IIIA mice treated with enzyme were still surviving.
Our results show that enzyme replacement from birth is effective in normalizing gait abnormalities observed in MPS IIIA mice and in reducing the amount of storage in various regions of the MPS IIIA mouse brain. A single dose of enzyme injected from birth is also effective in improving the reproductive outcome of MPS IIIA matings, and in increasing the life span of MPS IIIA mice.
brain of mice at various stages of their development.
MPS IIIA mice aged 6-, 12- and 18-weeks were injected with rhNS directly into two regions of the brain, the hippocampus and cerebellum (regions of the brain involved in memory and motor function, respectively). The brains of these mice were assessed at 24-weeks of age. Treatment was shown to reduce the amount of storage and other neurodegenerative changes in widespread areas of the MPS IIIA brain.
Manchester Children’s Hospital, Manchester, England
“Autologous Stromal Stem Cells as a target of genetic manipulation in the management of MPSII (Hunter syndrome)”
Stromal stem cells can be easily taken from bone marrow, expanded in the laboratory and differentiated into cartilage and bone cells. They have been shown to be able to participate in the repair of bone fracture and correct inherited skeletal disorders such as Osteogenesis Imperfecta. Hunter’s syndrome or MPSII is caused by the absence of an enzyme called Iduronidate-2- Sulphatase (I2S). This enzyme is important for the degradation of glycosaminoglycans (GAGs). A lack of this enzyme results in a multisystem disorder with developmental delay combined with bone and joint disease. Very limited benefit follows hematopoietic stem cell transplantation of MPSII, especially with regard to the skeletal disorder. Enzyme replacement therapy is under evaluation at present but seems to present a similar outcome with regard to the skeletal disease.
In this project we proposed to use stromal stem cells as target cells to correct the enzyme defect in Hunter’s syndrome, particularly in the skeleton as these cells could take part in the bone formation. We wanted to test the hypothesis that stromal stem cells from Hunter’s Syndrome could be corrected using a retroviral vector containing the I2S gene, and that the genetically modified stromal stem cells could generate bone expressing this transgene.
So far we have isolated stromal stem cells from two MPSII patients. The cells were able to expand in culture and differentiate into bone cells in a similar way to stromal stem cells isolated from normal donors. We have modified stromal stem cells from both MPSII patients and normal donors using a retroviral vector containing a marker gene called Green Fluorescent Protein (GFP). Cells containing this gene will appear green when exposed to a fluorescent light allowing for quantification. Over 90% of stromal stem cells from MPSII patients could be modified to produce GFP in a similar way to cells from normal donors. However introduction of the I2S gene into stromal stem cells of MPSII patients using a similar retroviral vector to the one used to produce GFP resulted in very poor correction of the enzyme with levels at best of 2.7% of normal. We hypothesized that this was due to the large size of the I2S gene, which made it more difficult to produce enough viral particles for efficient correction of the stromal stem cells. We have designed a new vector that can produce both genes, GFP and I2S, at the same time. Cells that produce GFP can be seen and separated from the rest. Once the separation has occurred we expected all of those cells to produce I2S. Indeed with this system I2S corrected marrow stem cells from MPSII patients showed levels of I2S enzyme 18 times higher than marrow stem cells from normal donors. Our visiting scientist from Brazil, who started working on this project in February, is now in the process of measuring levels of GAGs after selection of the cells and
correction of the enzyme deficiency. She is also looking at the ability of the corrected stromal stem cells to produce and secrete the I2S enzyme to correct the surrounding cells.
Parallel studies investigating the best conditions for transplantation of the stromal cells in the bone are also ongoing. These studies should enable us to assess the potential of using genetically modified marrow stromal cells for the therapy of skeletal disorders in MPSII patients by the end of the year.
In July 2002, The National MPS Society awarded three research grants. The recipients receive $30,000 for each of the two years of funding. Following are the reviews of the first year of research:
Dr. Douglas Brooks, Lysosomal Diseases Research Unit, Department of Chemical Pathology, Women’s and Children’s Hospital, North Adelaide, South Australia
New diseases involving proteins that control vesicle traffic within cells, are currently being recognized and many display a similar clinical presentation to that seen in lysosomal storage disorder patients. A new group of molecules called GGA proteins (Golgi localized, gamma ear containing, ARF binding proteins) have been shown to be involved in vesicle traffic within cells and interact with the mannose-6-phosphate receptors that normally target and transport lysosomal proteins to lysosomes. In this project, we planned to investigate patients characterized as having either I-cell disease or MLIII. Some of these patients will obviously have a deficiency in the enzyme N-acetylglucosamine 1-phosphotransferase that has been shown to cause I-cell disease. However, a number of these patients may also represent a disorder that has not been previously described. We hypothesized that a defect in one of the GGA proteins should give an identical clinical presentation to I-cell disease, based on the inability to correctly traffic mannose-6-phosphate receptors. We proposed two specific project initiatives: 1. Develop
monoclonal antibodies to specific regions on GGA proteins to allow the study of the cell biology and intracellular traffic of normal and mutant GGA proteins. 2. The characterization of I-cell and MLIII patients to identify potential defects in GGA proteins and vesicle traffic. Progress in the first year of this project has been excellent.
Development of monoclonal antibodies to specific regions on GGA proteins: There are three known GGA proteins. We have generated a panel of 22 monoclonal antibodies with reactivity to GGA1 (6), GGA2 (10) and GGA3 (6). All of the antibodies show specific reactivity to the respective GGA proteins and at least one monoclonal antibody from each set, detects denatured protein and shows reactivity on Western blots. Several of the monoclonal antibodies have had the positional location of the antibody reactivity mapped on the respective GGA protein (epitope mapped). We have also generated specific polyclonal antibodies to each of the three GGA proteins. These antibodies will allow the development of immunoquantification assays for determining intracellular levels of normal and mutant GGA proteins and facilitate a more sophisticated functional analysis of the different domains of these proteins. Preliminary analysis of the monoclonal antibodies has shown reactivity in immunofluorescence experiments and this should allow the proposed high resolution confocal imaging of cells from “I-cell” patients. In the second year of this project we plan to investigate the cell biology of GGA proteins in I-cell patients and identify patients with potential defects in GGA proteins.
Dr. Briony Gliddon, Lysosomal Diseases Research Unit, Department of Chemical Pathology, Women’s and Children’s Hospital, North Adelaide, South Australia
Two major experiments were included in the proposal,
Progress on this study has been very good, with most of the preparatory work now complete. Based on successful results with enzyme replacement therapy (ERT) of 1mg/kg rhNS intravenous injections into mucopolysaccharidosis type IIIA (MPS IIIA) mice from birth, it was decided to follow on from these experiments using enzyme doses 0.2, 1.0 and 5.0 mg/kg recombinant human sulphamidase (rhNS) to determine which dose gives greatest efficacy.
Before enzyme can be used for therapy in the mice it is necessary to produce and purify large amounts of enzyme to a high quality suitable for injection into mice. This has now been achieved. We have also developed various tests to evaluate and compare the behavior and learning abilities of MPS IIIA and unaffected control mice and therefore critically assess the efficacy of ERT in MPS IIIA mice. Male MPS IIIA mice display an aggressive behavioral phenotype from 8-12 weeks of age. It was observed in the preliminary experiment whereby 1mg/kg rhNS was injected into MPS IIIA mice from birth that this aggressive phenotype was delayed, a positive result of the therapy. At that stage we had no test to quantify this aggression in the mice and it remained only qualitative. Consequently we are designing a test known as the resident-intruder test, which is commonly used to measure aggression in mice. This test will be employed in the dose-dependent ERT experiment. A large number of mice (80 MPS IIIA and 40 unaffected) are needed for this study to make the behavioral testing statistically significant. This requires an extensive breeding program to obtain such a large number of mice, in which it is necessary that they be age matched (ie born at least in the same week). Breeding has been ongoing throughout enzyme purification and behavioral test analysis and we are nearing our final round of breeding whereby injections will commence into the mice.
The study described in the grant has been completed. The aim of this study was to determine whether enzyme replacement directly into the CNS of adult mice can prevent, delay or even reverse the functional deficits and pathological features of MPS IIIA in mice. In preliminary experiments one group of mice received enzyme treatment through intracerebral injections into both the hippocampus and cerebellum, (the sites related to pathology and behavior deficits in MPS IIIA mice) at 6 weeks of age, the other at 12 weeks of age and another at 18 weeks of age. All animals were sacrificed at 24 weeks of age. In both 6 and 12 week treated animals, but not 18 week treated mice, enzyme treatment reduced the formation of axonal spheroids, anomalies found in the diseased brains of untreated MPS IIIA mice. This suggests that spheroids which seem to form between 12-18 weeks of age are irreversible. Enzyme treatment also abolished the neurodegenerative changes seen in the retrosplenial cortex of untreated MPS IIIA mice. The observation of a reduction in the number of lysosomal storage vacuoles in treated mice appeared to be related to time post treatment, where mice treated most recently ie: at 18 weeks old at treatment, 24 weeks old at sacrifice showed the greatest reduction in storage vacuoles. This suggests clearance of storage by enzyme occurs rapidly but that in the absence of continued enzyme delivery, storage vacuoles reappear. These preliminary results are very encouraging. We
Dr. Robb Wynn, Consultant Hematologist, Manchester Children’s Hospitals, Manchester, England
We are grateful for the opportunity to report on the progress we have made during the first year of this two year grant from the National MPS Society Inc. We believe we have made very considerable progress in following the schedule of research as laid out in our original research schedule, and therefore towards achieving the aims of our research.
Expansion and differentiation of Mesenchymal Stem Cells (MSCs) from patients with MPSII We have attempted to do this in two ways in our laboratory as we had originally stated. We have used plastic adherence and compared the properties of MSCs from MPSII patients with MSCs from patients with other MPS and from age-matched controls. We have also attempted to isolate MAPCs (Multipotent Adult Progenitor Cells) as described by Reyes et al in Minneapolis. We have not been able to isolate MAPC in this manner ? these difficulties have been shared by scientific groups elsewhere (personal communication). The isolation and use of MAPCs have in this last year been patented by the Minneapolis investigators and we have therefore concentrated on MSCs isolated by plastic adherence.
We have obtained MSCs from MPS II patients, although the number of patients has been less than anticipated as participation in ERT renders the patient ineligible for marrow donation, according to the terms of those trials. Our Research Ethics requires that the marrow be done under general anaesthesia during significant other surgery.
We do demonstrate that:
We have also observed that the differentiation properties of the MPS II marrow are the same as age-matched MSCs. Taken together these data are consistent with our underlying hypothesis that MPS II MSCs are suitable targets for genetic manipulation.
We have commenced work on what we had originally scheduled for the second year of the grant. We have confirmed that MSCs from MPS II patients are as good a target for genetic manipulation as other MSC clones in our hands. We have used A GFP vector (fig 4, below) to obtain the same transduction frequency (up to 80-%).
We have gone on to prepare retroviral packaging cell lines and we have screened several of these clones by measurement of the IDS enzyme. We have several clones that have enzyme activity above the control we are continuing this process. MSCs of the MPS II patients will then will be exposed to retrovirus and enzymatic correction of the deficient tissue obtained.
In this year we plan to consolidate the research achievements of our first year. We have appointed a Brazilian scientist to join us for one year. We hope to have some more original marrow from MPS II patients to confirm our preliminary observations that the numbers and properties of these cells in MPSII is the same as that in age-matched controls. We will optimize the packaging cell line and transduction of MPS II MSC cells and confirm in vitro correction of the condition. In parallel we have other projects that are examining the homing of these cells to the sites where they will be required (CNS, bone) as well as their functional potential in vivo in models of MPS disease.
We are grateful to the Society for giving us the opportunity to pursue this research and, as we continue our studies, we hope to publish our work to allow others to learn from what we have achieved.
Annual report on the grant: “Evaluation of Neonatal Gene Transfer in Dogs and Cats with MPS” funded in 2003 in a joint venture by the Ryan Foundation and the National MPS Society to Dr. Mark Haskins at the University of Pennsylvania and Dr. Katherine Parker Ponder at Washington University.
Thank you,
Mark Haskins VMD, PhD Ponder, MD Professor
Pathology and Medical Genetics School of Veterinary Medicine
University of Pennsylvania
Katherine Parker Associate Professor
Internal Medicine, Biochemistry and Molecular
Biophysics, and Genetics Washington University School of Medicine
2001 Research Grant PDF Download
“A Proposal to Develop a Canine Model of MPS IIIB”. Mark Haskins, VMD, PhD, and Matthew Ellinwood, DVM, PhD, School of Veterinary Medicine, University of Pennsylvania.
A naturally occurring canine form of MPS IIIB was identified in two Schipperke dogs, whose cases had been referred to the University of Pennsylvania. Realizing the importance of having a large animal model of this condition, we applied to The National MPS Society, and received funding to characterize the genetics and pathology of this canine model, and to establish a research colony of these dogs.
We have completed the initial characterization of the pathology seen in the first two cases of this disease and a manuscript is in preparation for submission to the Journal of Inherited Metabolic Disease. The clinical findings in canine MPS IIIB included a severe loss of balance, which began in early adulthood. Dogs also began to lose weight as the disease progressed. Both dogs were eventually euthanized because of the severe and progressive loss of balance. Analysis of these animals showed prominent GAG storage in the liver and kidneys of these dogs. The cerebellum, that part of the brain which governs balance, was severely affected, with a marked loss of neurons. Other parts of the brain showed marked neuronal storage, with storage also evident in macrophage-like cells. For the most part, the rest of the organs in the body had storage that was limited to macrophages. All tissues tested had a marked decrease of N acetyl-a-D- glucosaminidase (NAGLU) activity, to about 5% of normal. In tissues with severe storage, there were elevations seen in other enzymes, including b-glucuronidase and total b-hexosaminidase.
We have now isolated and sequenced the entire protein-coding region of the canine NAGLU gene. Our sequence extends from position -7 from the first ATG (in the context of a strong initiator site from -3 to +4), to the polyadenylation signal. The open reading frame of 2244 nucleotides bears substantial identity to human (87%) and mouse (84%) NAGLU sequences. The predicted protein comprises 747 amino acids, again with considerable identity to the human (83%) and mouse (79%) proteins. The amino terminus contains a stretch of 23 hydrophobic amino acids consistent with a signal peptide. The predicted mature protein, minus any possible carbohydrate residues, comprises 724 amino acids with a mass of 81.2 kDa. These data are due to be presented at the American Society of Human Genetics, in October of 2002.
The identification of the normal canine NAGLU sequence will be critical in developing this model as a means to evaluate enzyme replacement and gene therapies. The normal NAGLU gene sequence will allow us to next identify the disease causing mutation, and develop a DNA-based diagnostic, to be used to manage the research colony, and to help identify carrier animals in the pedigreed Schipperke population.
During the first year of this grant award we have produced two litters of pups, both sired by a Schipperke dog that is a carrier for canine MPS IIIB. Our breeding population in the colony is now two carrier males and four carrier females. We have three pregnant females at this time, with pups due in late June and July of 2002. We expect up to 8 litters in the coming year. Pups with MPS IIIB will be monitored very closely to document all early clinical signs and evidence of disease pathology.
The preliminary characterization of this model was presented at the American Society of Human Genetics meeting in San Diego in October of 2001, with acknowledgements to The National MPS Society for funding support (Ellinwood N.M., Wang P., Skeen T., Sharp N., Cesta M., Bush W., Hardam E., Haskins M.E., Giger U. (2001) Characterization of a canine model of mucopolysaccharidosis IIIB. American Journal Of Human Genetics. 69, Suppl. 482).
Engraftment of human hematopoietic cells leads to widespread distribution of donor-derived cells and correction of tissue pathology in a murine xenotransplant model of lysosomal storage disease.
A.A. Hofling1, C. Vogler2, M. Creer2, M.S. Sands1
Washington University School of Medicine, St. Louis, MO; St. Louis University School of Medicine, St. Louis, MO.
Bone marrow transplantation (BMT) is a relatively effective form of therapy for some lysosomal storage diseases. However, BMT has limitations including the harsh conditioning regimens (irradiation or cytotoxic drugs) required for engraftment and the possibility of life threatening immune reactions due to donor incompatibility. Some of the immune complications may be overcome by genetically modifying (gene therapy) the affected patient’s own bone marrow cells outside the body (ex vivo). These genetically modified cells now produce the deficient enzyme and can be transplanted back into the patient where they can give rise to all of the blood cells.
This gene therapy approach has proven effective in some mouse models of lysosomal storage disease. However, in those experiments mouse bone marrow cells, not human cells, are genetically modified to produce the deficient enzyme. Unfortunately, the gene therapy techniques used in the mouse studies have proven ineffective in human bone marrow cells.
Therefore, alternate gene therapy techniques need to be developed and tested directly in human cells. In order to accomplish this goal we recently developed a mouse model of mucopolysaccharidosis type VII (MPS VII, b-glucuronidase deficiency) that is capable of accepting human bone marrow cells. The transplantation and engraftment of cells from one species (human) into another (mouse) is referred to as xenotransplantation. When normal human bone marrow cells are injected into the xenotransplantation model of MPS VII they repopulate the blood system, and bone marrow-derived cells can be found in most tissues of the body. The number of human cells and level of enzyme activity in the transplanted mice is sufficient to reduce or nearly eliminate the lysosomal storage in many tissues. This xenotransplantation mouse will now allow us to test new gene therapy approaches directed at human bone marrow cells within the context of an authentic model of lysosomal storage disease. We have preliminary data showing that we can genetically modify human bone marrow cells and transplant them into the mouse xenotransplant model. The genetically modified human cells are producing a marker gene (non-therapeutic) which is easy to follow in the mouse. We have recently developed a gene therapy vector containing the b-glucuronidase gene (therapeutic) and are currently testing this in the mouse model. This type of experiment will provide important pre-clinical data before these novel approaches are attempted in affected patients.
Yoshio Nishimura* and Jeffrey D. Esko**
*Institute of Microbial Chemistry, Japan and ** University of California at San Diego, USA
Current therapies for the treatment of the mucopolysaccharidoses focus on methods of increasing enzyme concentration in patients to compensate for the enzyme deficit. Lowering the rate of biosynthesis of mucopolysaccharides should decrease accumulation of this substrate. The goal of any substrate deprivation therapy is not inhibit biosynthesis completely but to reduce this capability to levels commensurate with normal growth and development. We have investigated iminosugar inhibitors of 2-0-sulfotransfersases and N-deacetylase/N-sulfotransferase in mucopolysaccharide (glycosaminoglycan, GAG) biosynthesis directed towards agents for Hunter (deficient of iduronate 2-0-sulfatase) and Sanfilipo (deficient of N-acetylglucsamine N-sulfatase) syndromes. We have synthesized 34 gem-diamine 1-N-iminosugars as substrate analogs based on the structure of the constituent carbohydrates involved in heparan sulfate, one glycosaminoglycans (GAG). They resemble iduronic acid of N-acetylglucosamine, the acceptor sugar moieties acted upon by 2-0-sulfotransfersases or N-deacetylase/N-sulfotransferase, respectively. We considered conversion of functional groups and/or stereochemistry of gem- diamine 1-N-imosugars corresponding to the above acceptor sugar moieties (iduronic acid and N-acetylglucosamine) as an initial probe of structure – activity profile. These compounds contain the different functional groups and/or stereochemistry at positions of sulfation from the original sugar moieties of heparan sulfate, and are also alkylated by the various fashions at position linked to the adjacent sugar moieties in heparan sulfate for binding more avidly to the sulfotransferases. These compounds will be evaluated for inhibitory activity against heparan sulfate 2-0-sulfotransfersases and heparan sulfate N-deacetylase/N-sulfotransferase Initial studies will focus on determining the apparent Km values for the natural acceptor. The various inhibitors will then be tested at different concentrations to determine relative inhibitory constants.
Based on the biological data, we will further study on the structure-activity relationship of 1-N- iminosugars directed toward a candidate for substrate deprivation therapy of MPS.
Anne K. Hennig, Ph.D., is a 2001 recipient of an MPS Society two-year research grant for her project “In Utero Gene Therapy for Mucopolysaccharidoses”. Dr. Hennig works with Dr. Mark Sands at Washington University School of Medicine in St. Louis, MO. Following are the findings from the second year of her grant.
The proposal I originally submitted was for investigating “In Utero Gene Therapy for Mucopolysaccharidosis,” but because of technical difficulties associated with injecting the gene therapy into mouse fetuses that project is behind schedule and I do not yet have results beyond those I shared in the last progress report.
During the funding period I was also performing another series of experiments in which an identical adeno-associated virus (AAV) gene therapy vector was injected into the vitreous humor in the eyes of young adult MPS VII mice. These mice have no functional beta-glucuronidase (GUSB) and model Sly Syndrome. The objective of the study was to determine whether reducing lysosomal storage within the eye prevented retinal degeneration, and investigate what effect the treatment had on retinal function. We found high levels of GUSB enzyme expression within treated retinas 4 to 6 weeks after treatment, and the enzyme levels were still high 12 weeks after treatment, the endpoint of the study. Interestingly, GUSB activity also appeared in the optic nerve and the parts of the brain where axons from the injected eye travel. To try to determine whether this enzyme activity was being produced locally by brain cells that had been infected by the gene therapy viral vector, I looked for genetic material from the virus in small tissue samples from different sites showing enzyme activity. The PCR assay I used could detect a single virus genome in one cell among 2 -10 thousand uninfected cells. Testing tissue samples from three treated mice, I found evidence of viral DNA in the injected eyes but not in the optic nerves coming from those eyes, or in any of the brain regions that contained GUSB activity. This indicated that the GUSB enzyme in the brain was probably not being made by the brain cells in the areas where I saw the enzyme activity. Therefore, GUSB was most likely produced in the treated eye and transported along the optic nerve into the brain.
Brains from some of the treated animals were examined for lysosomal storage. Storage was reduced in the parts of the brain along the visual pathway where we had seen GUSB activity, and also in other visual areas farther away. This indicated that small amounts of enzyme, too little to detect with our staining assay, must have been being passed along the next set of connections in the visual pathway. What surprised us was that correction of lysosomal storage was also seen in other parts of the brain that were not involved in vision but happened to be located next to visual areas. The most exciting of these areas was the hippocampus, which stores and recalls memories as part of the learning process. Unfortunately, our treated MPS VII mice could not be given behavioral tests because their systemic disease had progressed too far (we only treated their eyes). It will be important to determine whether cognitive function might be restored by this approach.
A manuscript describing these findings has been accepted for publication in the Journal of Neuroscience later this month. In future experiments I am planning to extend these findings to MPS IIIB knockout mice. Since these mice have lysosomal storage pathology in the eye and brain without debilitating systemic problems, I believe that behavioral testing will be possible in order to investigate the impact of the treatment on learning and memory.
Use the table sort feature to find trials that best fit your interest. Please follow links to visit clinicaltrials.gov to learn more.
| Clinical Trial, Briefly Title and ClinicalTrials.gov link | Phase | Status | Sponsor reports | MPS Type | Interventions, Sponsor Study Name, &Treatment designation | Study Sites | Vector type | Route: ICM: Intracisternal magna IV: Intravenous IC: Intracranial |
|---|---|---|---|---|---|---|---|---|
| RGX‐111 Gene Therapy in Patients With MPS I | I/II | Recruiting | December 1, 2020 | Type I | REGENXBIO, RGX‐111 | Children’s Hospital of Orange County, Orange, California, United States Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, United States | AAV9 | ICM |
| Ascending Dose Study of Genome Editing by the Zinc Finger Nuclease (ZFN) Therapeutic SB‐318 in Subjects With MPS II | I/II | Active, not recruiting | February 7, 2019 | Type I | Sangamo Therapeutics, Empowers Study, SB‐318 | UCSF Benioff Children’s Hospital Oakland, Oakland, California, United States | AAV6 | IV |
| Gene Therapy With Modified Autologous Hematopoietic Stem Cells for the Treatment of Patients With Mucopolysaccharidosis Type I, Hurler Variant | Active, not recruiting | September 1, 2020 | Type I | Orchard Therapeutics, OTL‐203 | Ospedale San Raffaele, Milano, Italy | Second generation self‐inactivating lentiviral vector transduced autologous HSC | IV‐Transfusion | |
| RGX‐121 Gene Therapy in Patients With II (Hunter Syndrome) | I/II | Recruiting | December 9, 2020 | Type II | REGENXBIO, RGX‐121 | UCSF Benioff Children’s Hospital, Oakland, California, United States Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, United States Children’s Hospital of Pittsburgh ‐ UPMC, Pittsburgh, Pennsylvania, United States Hospital de Clinicas de Porto Alegre, Porto Alegre, RS, Brazil | AAV9 | ICM |
| RGX‐121 Gene Therapy in Children 5 Years of Age and Over With II (Hunter Syndrome) | I/II | Not yet recruiting | Type II | REGENXBIO, RGX‐121 | Not yet listed | AAV9 | ICM | |
| Ascending Dose Study of Genome Editing by the Zinc Finger Nuclease (ZFN) Therapeutic SB‐913 in Subjects With MPS II | I/II | Active, not recruiting | February 7, 2019 | Type II | Sangamo Therapeutics, Champion Study, SB‐913 | UCSF Benioff Children’s Hospital Oakland, Oakland, California, United States Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, Illinois, United States NYU School of Medicine, Neurogenetics Division, New York, New York, United States University of North Carolina, Chapel Hill, North Carolina, United States Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, United States | AAV6 | IV |
| Phase I/II Gene Transfer Clinical Trial of scAAV9.U1a.hSGSH | I/II | Recruiting | November 9, 2020 | Type IIIA | Abeona Therapeutics, The Transpher A Study, ABO‐102 | Nationwide Children’s Hospital, Columbus, Ohio, United States Women’s and Children’s Hospital, North Adelaide, South Australia, Australia Hospital Clinico Universitario de Santiago, Santiago De Compostela, Spain | AAV9 | IV |
| Gene Transfer Study of ABO‐102 in Patients With Middle and Advanced Phases of IIIA Disease | I/II | Recruiting | Type IIIA | Abeona Therapeutics, Study ABT‐003, ABO‐102 | Children’s Hospital of Pittsburgh, Pittsburgh, Pennsylvania, United States Adelaide Women’s and Children’s Hospital, North Adelaide, South Australia, Australia Hospital Clinico Universitario de Santiago, Santiago De Compostela, Spain | AAV9 | IV | |
| Gene Therapy With Modified Autologous Hematopoietic Stem Cells for Patients With Mucopolysaccharidosis Type IIIA | I/II | Recruiting | December 8, 2020 | Type IIIA | Orchard Therapeutics, OTL‐201 | Manchester University NHS Foundation Trust, Manchester, United Kingdom | Second generation self‐inactivating lentiviral vector transduced autologous HSC | IV‐Transfusion |
| Study of AAVrh10‐h.SGSH Gene Therapy in Patients With Mucopolysaccharidosis Type IIIA (IIIA) | II/III | Active, not recruiting | October 15, 2020 | Type IIIA | Lysogene, LYS‐SAF302 | CHOC Children’s, Orange, California, United States University of Minnesota, Minneapolis, Minnesota, United States Weill Cornell Medical College, New York, New York, United States Baylor college of medicine / Texas children’s hospital, Houston, Texas, United States Armand Trousseau Public Hospital, Paris, France University Medical Center Hamburg‐Eppendorf, Hamburg, Germany Amsterdam UMC, Amsterdam, Netherlands Great Ormond Street Hospital, London, United Kingdom | AAVrh10 | IC |
| Gene Transfer Clinical Trial for Mucopolysaccharidosis (MPS) IIIB | I/II | Recruiting | November 9, 2020 | Type IIIB | Abeona Therapeutics,The Transpher B Study, ABO‐101 | Nationwide Children’s Hospital, Columbus, Ohio, United States Armand‐Trousseau Hospital, Paris, France Hospital Clinico Universitario de Santiago, Santiago De Compostela, Spain | AAV9 | IV |
| Gene Therapy in Patients With Mucopolysaccharidosis Disease | I/II | Recruiting | No Results Available | Type VI | TIGEM, MESIX, AAV2/8.TBG.hARSB | Federico II University, Napoli, Naples, Italy Erasmus Medical Center, Rotterdam, Netherlands Children’s Hospital Hacettepe University, Ankara, Turkey | AAV8 | IV |
